The Cholesterol storage disease is a lysosomal storage disease and congenital metabolic disease with a genetic basis. The disease is hereditary and is caused by a genetic mutation in the genes coding for lysosomal acid lipase. The symptomatic treatment of the patient takes place conservatively with medication or through enzyme replacement therapeutic steps.
What is Cholesterol Ester Storage Disease?

© 4th Life Photography - stock.adobe.com
The group of lysosomal storage diseases comprises a number of congenital diseases that can be traced back to inadequate or defective activity of the so-called lysosomes. All diseases in the group are metabolic diseases. One such disease is the cholesterol ester storage disease, which is commonly called CESD is abbreviated.
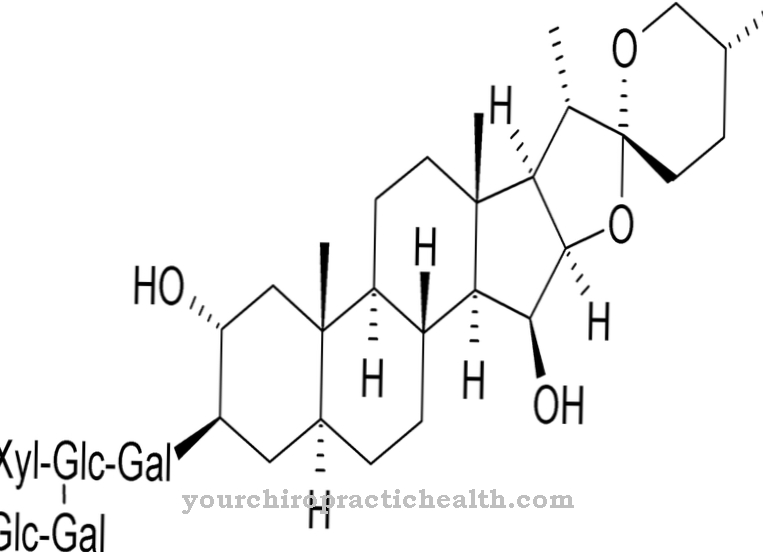
As with all lysosomal storage diseases, CESD is also due to a lack of activity of certain lysosomes. The lack of activity in this disease relates to the lysosomal acid lipase, which breaks down cholesterol esters and triacylglyceride in healthy people. Cholerestine ester storage disease is an extremely rare metabolic disease and is associated with heredity. In this context, inheritance corresponds to autosomal recessive inheritance.
The disease is one of the innate metabolic diseases. However, the first symptoms do not have to manifest themselves immediately after birth. The primary cause of the disease is a genetic mutation. A disease with a similar gene mutation is Wolman disease. In contrast to this disease, the cholesterol ester storage disease is characterized by a significantly milder course, since a residual activity of the lysosomal acid lipase is retained and the cholesterol ester breakdown can bring about at least outside the liver.
causes
Patients with cholerestine ester storage disease suffer from an enzyme defect in lysosomal acid lipase. This defect leads to a reduced breakdown of cholerest esters. This results in an accumulation of cholesterol esters and the just as little degraded triglycerides. The coding gene for acid lipase is located in the DNA on chromosome 10 in the gene locus q23.2 to 23.3. Ten exons make up the gene.
The cause of choleretine ester storage disease is a nonsense or missense mutation in the exons involved. Frame shifts or the skipping of certain exons can also be the cause. The mutated gene product shows reduced activity, so that hardly any lipids of the lysosome can penetrate into the cytoplasm. In this way, the control loop for regulating intracellular cholesterol concentrations is interrupted.
An intracellular low cholesterol concentration arises and brings the endogenous cholesterol synthesis and the LDL receptor activity to an upregulation. As a result, the lysosome takes up endocytosed cholesterol. Due to the endogenous synthesis of cholesterol, the cells are overloaded and lipid vacuoles form. The vacuoles lead to a loss of function of the individual cells, trigger fibrosis and cause cell death.
You can find your medication here
➔ Medicines for painSymptoms, ailments & signs
Cholerestine ester storage disease is characterized by a characteristic complex of clinical symptoms. Liver abnormalities are particularly typical abnormalities. As an organ, the liver is significantly involved in the metabolism of cholesterol, which explains the preferred first manifestation of the disease in the liver. In patients with cholerestine ester storage disease, the symptoms manifest themselves in most cases much later than in patients with Wolman disease.
In the latter, the first symptoms usually manifest themselves more or less immediately after birth. In contrast, patients with cholerestine ester storage disease can remain asymptomatic for a long time. The main symptom is hepatomegaly. The liver swells up and develops into a fatty liver as the deposits progress. In addition, hypercholesterolemia occurs in most cases.
This hypercholesterolemia corresponds to a lipid metabolism disorder due to increased cholesterol level in the blood. These symptoms are usually associated with hyperlipidemia and a reduced HDL concentration. In addition to the swelling of the liver and the loss of function, most of the symptoms are exclusively diagnosable by laboratory diagnosis.
diagnosis
For the diagnosis of cholerestine ester storage disease, a laboratory blood analysis is required, which can detect the changed lipid pattern and document any foam cells that occur. In addition, a liver biopsy can be performed as part of the diagnosis, which shows massive lysosmal accumulations.
In terms of differential diagnosis, the disease must be differentiated from other lysosomal storage diseases within the framework of diagnosis. This distinction is usually made in the context of enzymatic activity tests. The genetic tests to identify the mutation are rarely used to differentiate.
Complications
Cholesterol ester storage disease can lead to various complaints and complications, which primarily depend on the severity of the disease. In most cases, however, the liver is affected. In many patients, the first signs of cholesterol ester storage disease appear immediately after birth, so that, for example, the liver is swollen or if it later develops into a so-called fatty liver.
Most often, patients also experience pain and a stinging from the swelling. Diagnosis is usually relatively easy with a blood test, so there is no delay in diagnosis. Unfortunately, a causal therapy and treatment of the cholesterol ester storage disease is not possible, so that above all the symptoms have to be limited.
This results in a reduced absorption of cholesterol. However, the patient is dependent on taking medication for the rest of his life. Otherwise the cholesterol ester storage disease will not cause any further complaints or complications. Life expectancy is also not reduced with therapy. The patient's everyday life is rarely restricted by the disease. When planning children, however, the likelihood of worshiping the disease should be examined.
When should you go to the doctor?
Cholesterol ester storage disease is usually diagnosed immediately after birth. Typical signs that point to the disease and that need to be clarified and treated are a swollen liver and, as the disease progresses, signs of fatty liver disease. If the child complains of sharp pain in the liver, the pediatrician should be consulted immediately. This is especially true if there are additional complaints that indicate the cholesterol ester storage disease.
In the event of severe complications, the emergency medical service should always be contacted. Although life-threatening symptoms rarely occur with the disease, chronic liver disease can develop. Therefore, at the first signs of cholesterol ester storage disease, you should speak to a doctor.
If there are already known cases of the disease in the family, an examination immediately after the birth is recommended. Under certain circumstances, the disease can also be diagnosed prenatal. The doctor will then discuss with the parents which further measures are possible as part of a treatment.
Doctors & therapists in your area
Treatment & Therapy
The cholerestine ester storage disease is due to a genetic defect. Therefore, no causal therapeutic steps for treating the patients are available to date. A causal therapy would only be possible in the context of gene therapy approaches. Gene therapy has not yet reached the clinical phase. Therefore, the storage disease is still considered incurable today and is treated only symptomatically. Symptomatic therapy focuses on a reduced uptake of cholesterol.
For this purpose, the intestinal cholesterol absorption of the patient is conservatively inhibited with medication. Drugs such as cholestyramine and ezetimibe are suitable for inhibition. In addition, patients usually receive statins, which inhibit HMG-CoA reductase. In the recent past, new treatment options have also been established, especially enzyme replacement therapies.
For patients with cholerestine ester storage disease, the enzyme sebelipase alfa plays a major role.Enzyme replacement therapies with this enzyme are currently used in clinical trials and were approved and positively evaluated by the European Medicines Agency last year.
Outlook & forecast
Cholesterol ester storage disease has an unfavorable prognosis. The disease is considered incurable and can lead to difficult complications. The recessive hereditary disease is treated symptomatically by doctors, since for legal reasons, interference with human genetics is currently not permitted.
The severity of the disease is individual and therefore different for each patient. Accordingly, there is also no uniform treatment plan. Once the liver is compromised, the prospect of health improvement drops significantly. A swelling of the liver or a fatty liver can lead to further illnesses. There is often more inflammation or cirrhosis of the liver. At this stage, it is almost impossible to alleviate the symptoms. The sick person is threatened with liver failure and thus premature death.
Patients whose liver does not suffer any damage are in significantly better health. As long as they follow the guidelines of the doctors and avoid the intake of products containing cholesterol in addition to medication, they experience a good quality of life despite the storage disease.
Since the treatment is a long-term therapy, the state of health deteriorates within a short time as soon as the drug is discontinued. An unhealthy lifestyle also has an immediate negative impact on the patient's well-being.
You can find your medication here
➔ Medicines for painprevention
Cholerestine ester storage disease is a genetic disease. Since no external factors are known to be causative factors, the disease can only and so far only be prevented through genetic counseling in the family planning phase. However, this also cannot rule out new mutations.
Aftercare
In most cases, there are no special follow-up measures available to those affected with cholesterol ester storage disease. The patient is primarily dependent on a quick diagnosis so that the symptoms can be properly alleviated, as this cannot lead to self-healing. Therefore, the person concerned should contact a doctor as soon as the first symptoms and complaints appear in order to prevent the symptoms from worsening.
Since this is a genetic disease, a genetic examination and consultation should always be carried out if you want to have children in order to prevent the cholesterol ester storage disease from occurring again. In most cases, the disease is treated with medication.
The person affected should ensure that they are taken regularly and that the dosage is correct so that the symptoms are permanently alleviated. Consult a doctor first if anything is unclear or if you have any questions. With cholesterol ester storage disease, many patients need help and support from friends and family. Contact with other people affected by the disease can also be very useful.
You can do that yourself
Cholesterol ester storage disease is a genetic disorder. Therefore, there are currently neither conventional nor alternative methods to treat the cause of the disease. Accordingly, there are no self-help measures available that combat the disease causally.
However, patients can still make an important contribution to avoid the progression of the disease and severe consequential damage, especially to the liver. A low-cholesterol diet is of central importance. Patients should obtain comprehensive and competent information about the special diet requirements of the cholesterol ester storage disease.
The attending physician is not always the best possible contact, as nutritional issues still hardly play a role in the training of physicians. Those affected should therefore consult a nutritionist or an ecotrophologist.
In general, cholesterol is only found in foods of animal origin. Switching to a vegan diet therefore makes sense for those affected. In any case, foods of animal origin, which are particularly high in cholesterol, should be avoided.
These include in particular fatty meat, sausage products, offal, eggs, butter, cream and whole milk. Eggs are often consumed hidden. Patients are often unaware that many pasta products, especially pasta and pastries, as well as ready meals and mayonnaise, contain large amounts of eggs and correspondingly high amounts of cholesterol.