The Holt-Oram Syndrome is a malformation syndrome that is primarily associated with heart defects and anomalies of the thumbs and is caused by a mutation. In most cases, the causative mutation occurs sporadically and thus corresponds to a new mutation. The surgical correction of the heart defect is the focus of the therapy.
Holt-Oram Syndrome?

© SmirkDingo - stock.adobe.com
The congenital malformation syndromes with predominant involvement of the extremities are a group of diseases that includes various deformities of the arms and legs. One of them is this Holt-Oram Syndrome, also called Heart-hand syndrome is known. The syndrome is associated with the group of atriodigital dysplasias, which include congenital diseases with malformations of the upper extremities and the heart.
In addition to the Holt-Oram syndrome, the heart-hand syndrome type 2, the heart-hand syndrome type 3 and the heart-hand syndrome of the Slovenian type belong to this group of diseases. The Holt-Oram syndrome was first described in 1960. The British pediatrician Holt and the cardiologist Samuel Oram are the first to describe the disease. The Holt-Oram syndrome This is a comparatively rare disease and is associated with an average prevalence of one affected person in 100,000 people.
The cause of the malformations of the hands and the heart in Holt-Oram syndrome lies in genetics. The symptoms of the syndrome are as varied as its suspected causes. The malformations in the disease concentrate on the heart and thumb.
causes
Although Holt-Oram syndrome is a genetic and congenital disease, there is hardly any familial frequency in the cases documented so far. Although the syndrome seems to be inherited in an autosomal dominant mode of inheritance in individual cases, a large part of the case documentation suggests its sporadic occurrence. Around 85 percent of the documented cases seem to be due to a new mutation.
The primary cause of the Holt-Oram syndrome lies in genetic mutations at the gene locus 12q23-24.1 At this gene locus is the so-called TBX5 gene, which is located on chromosome 12 and for a protein involved in the limb and limb Coded heart development. The exact functions of the protein have not yet been clarified. It has also not yet been established whether external factors such as exposure to toxins or malnutrition in the mother during pregnancy favor the mutation of the TBX5 gene.
Mutations in the gene can be detected in at least up to 70 people out of 100 patients with Holt-Oram syndrome. However, science assumes that abnormalities in other genes can also cause the symptoms of the syndrome. For example, malformation syndrome is associated with tripartite thumb polysyndactyly.
Symptoms, ailments & signs
Patients with Holt-Osram syndrome suffer from a complex of malformations that primarily affect the thumb and heart. Although the localization of the patient's malformations is common, different types of malformations on the heart and thumb are possible. The clinical picture is therefore extremely varied.
The cardiac defects can manifest themselves, for example, in the form of a ventricular septal defect, an atrial septal defect, a cardiac arrhythmia or a conduction disorder. The skeletal anomalies can correspond to reduction malformations of the thumbs, but anomalies such as the non-application of the spoke can also occur.
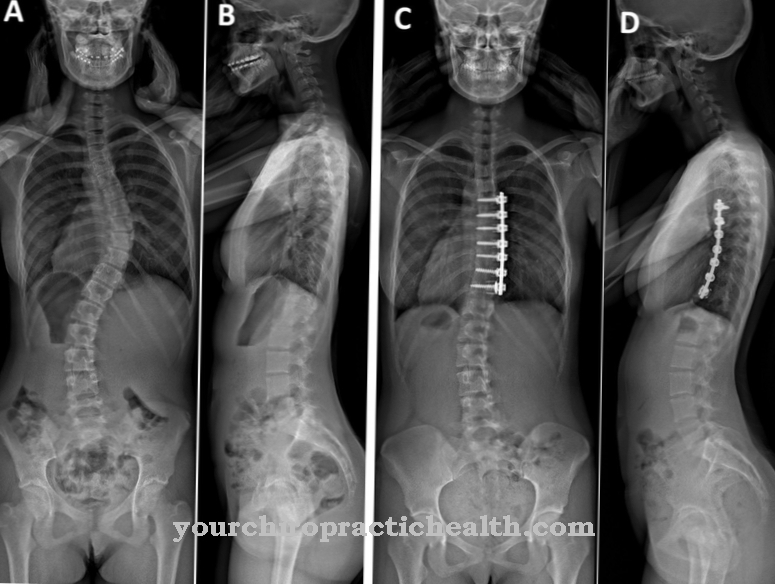
Many patients with the syndrome also suffer from radioulnar synostoses and abnormalities of the ribs, scapula, or clavicle. In addition, Holt-Oram syndrome is associated with pectus carinatum and scoliosis. The majority of those affected also suffer from syndactylia of the fingers or toe phalanges. In individual cases these symptoms are associated with hypertelorism.
Diagnosis & course of disease
Holt-Oram syndrome is often misdiagnosed. In a differential diagnosis, the doctor must differentiate the symptom complex from Okihiro syndrome after gene mutations in the SALL4 gene on chromosome 20, which is associated with the same arm malformations and heart defects. What is particularly relevant in the differential diagnosis is that patients with Okihiro syndrome usually have a Duane anomaly.
They squint, are often affected by kidney malformations and have hearing disorders, foot anomalies or ear malformations. The thrombocytopenia-absent-radius syndrome must also be differentiated from the Holt-Oram syndrome, which is mainly achieved through laboratory diagnostics. Other clinical pictures with a clinically similar picture are Fanconi anemia and Pallister Hall syndrome.
The life expectancy of patients with Holt-Oram syndrome is not below average. Only in severe cases is there a hardly treatable heart defect that makes the prognosis unfavorable.
Complications
Holt-Oram syndrome causes a number of different malformations and deformities in the patient, which can make life and everyday life difficult. Above all, the heart is affected by the malformations, so that the patient suffers from a heart defect. There is also a cardiac arrhythmia from which the person affected can die in the worst case.
Anomalies also occur on the thumbs, so that certain movements or processes in everyday life are also made more difficult. It is not uncommon for the deformities on the body to lead to teasing and bullying of other children, which can lead to psychological complaints and depression in many patients. It is not uncommon for kidney disorders to occur, which in the worst case can lead to renal insufficiency.
Furthermore, those affected also suffer from a visual impairment and a hearing impairment. As a rule, life expectancy remains unchanged as a result of Holt-Oram syndrome, as long as there is no heart defect that leads to death. A causal treatment of Holt-Oram syndrome is usually not possible, so only the symptoms can be treated. In many cases, psychological support is also necessary.
When should you go to the doctor?
Holt-Oram syndrome is usually diagnosed shortly after birth. Depending on how severe the malformations are, the affected child may need further medical examinations. In principle, the cardiac defects must be treated promptly in order to reduce the risk of serious secondary diseases. If complications such as cardiac arrhythmias or signs of atrial septal defect develop, medical advice is required. A medical professional should also be consulted with skeletal thumb abnormalities.
The parents of affected children should consult their doctor closely and inform them of any unusual symptoms. Since Holt-Oram syndrome is a hereditary disease, no causal treatment is possible. The patients may therefore have to be treated for a lifetime, depending on which malformations occur and what the patient's constitution is. As this often also causes emotional complaints, accompanying psychological support is indicated. Children who suffer from bullying or teasing should seek therapeutic counseling with their parents.
Doctors & therapists in your area
Therapy & Treatment
No causal treatments are available for patients with Holt-Oram syndrome. There is hope for a causal treatability in the future, since gene therapy is currently the subject of medical research. However, as long as this type of therapy does not reach the clinical phase, Holt-Oram syndrome remains an incurable disease.
At the moment, only symptomatic therapy options are available for treating patients. The therapy is based on the symptoms in the individual case. The early correction of the heart defect is of particular relevance. This correction is done surgically. In the case of a Vohof septal defect, the surgical procedure aims to close the defect in question, for example. The same is true for a ventricular septal defect.
Corrections to the malformations on the extremities are initially of secondary importance. After successfully correcting the heart defect, reconstructive surgical procedures can restore missing spokes and separate syndactylies. An existing scoliosis is usually fought under physiotherapeutic care. In particularly severe cases, surgery to implant a titanium rib prosthesis may be necessary.
In most cases, no intervention is required for the pectus carinatum. For psychological reasons, however, the thorax can be reshaped surgically, following the Nuss procedure, for example.
Outlook & forecast
The prognosis for Holt-Oram syndrome is favorable. Although there is a genetic defect, it can be adequately treated with current medical options. The life expectancy of someone with the syndrome is in most cases not below the average. In the case of a severe malformation, there can be significant losses in life expectancy. The prognosis is clearly worsened in these patients. The heart's activity is restricted and can lead to a premature end of life.
However, the majority of patients can be treated well and successfully. Although there is no cure due to the genetic defect present, there are good prospects for various correction options. The heart's activity is regulated and, if possible, completely corrected in a surgical procedure. Although there can be a permanent impairment in the way of life in comparison to healthy people, a good quality of life is achieved due to the treatment.
Physical abnormalities or malformations are changed in a further step. Usually, after the child's growth phase is complete, a necessary or desired correction of the existing malformations is initiated. If the disturbances in the development process lead to significant impairments, corrective measures are taken in childhood to alleviate the symptoms. Due to the optical changes, the patient may experience psychological consequences. This worsens the overall prognosis.
prevention
So far the Holt-Oram syndrome cannot be prevented because the external influencing factors have not been conclusively clarified.
Aftercare
Since Holt-Oram syndrome is a congenital disease, it cannot be completely cured. Therefore, the measures or the possibilities of follow-up care are very limited, so that the person affected is primarily dependent on early detection and subsequent treatment. If the patient or the parents want to have children, genetic counseling is given to prevent this syndrome from occurring again.
The treatment of Holt-Oram syndrome is primarily aimed at treating the heart defect. This is corrected by a surgical procedure, whereby the patient must recover and rest after the procedure. Exertion or physical activity should be avoided. It is not uncommon for physiotherapeutic treatment to be carried out, whereby many of the exercises can also be performed in your own home.
Those affected are sometimes dependent on the help and support of their own family and friends. This can also prevent psychological upsets or depression. Furthermore, a healthy lifestyle with a healthy diet has a very positive effect on the course of the Holt-Oram syndrome.
You can do that yourself
The Holt-Oram syndrome cannot be prevented and also cannot be treated with means of self-help. With this syndrome, those affected are always reliant on an operation to treat the heart defect in order to extend the patient's life expectancy. The earlier the syndrome is recognized, the higher the chances of a positive course of the disease. The other malformations on the body must also be corrected surgically.
Since many of those affected also suffer from psychological complaints or from inferiority complexes associated with this syndrome, they are dependent on psychological treatment. However, conversations with other patients, your own parents or friends can also strengthen the patient's self-confidence and thus alleviate the psychological complaints. Furthermore, some patients are dependent on the help of their fellow human beings in their everyday life, whereby warm care has a very positive effect on the course of the Holt-Oram syndrome.
Since the disease can also affect the internal organs, the patients are dependent on regular examinations and controls by various doctors. This can prevent kidney problems, for example. Affected children should be informed about the consequences and complications of the disease.

.jpg)

.jpg)







.jpg)




.jpg)