The Mucopolysaccharidosis is a collective term for lysosomal storage diseases that are based on the storage of glycosaminoglycans. All diseases develop similar symptoms and forms. The severity of the syndromes varies greatly.
What is mucopolysaccharidosis?

© Sebastian Kaulitzki - stock.adobe.com
A Mucopolysaccharidosis there is no such thing as a single disease. The term mucopolysaccharidosis is a collective term for a large number of storage diseases. These are based on storage disorders of glycosaminoglycans (GAG) in the lysosomes of cells. The storage takes place progressively because the termination of the connections does not work.
All mucopolysaccharidoses are genetic. Every disease lacks a specific enzyme that catalyzes the breakdown of the corresponding GAG. All mucopolysaccharidoses are very rare diseases and often show similar courses. If left untreated, the ever-increasing deposits destroy the cells. The organs are destroyed in the process. The disease can begin in infancy as well as in adulthood.
Mucopolysaccharidosis can be caused by four different groups of glycosaminoglycans:
- Heparan sulfate
- Keratan sulfate
- Chondroitin sulfate
- Dermatan sulfate.
All glycosaminoglycans consist of a polysaccharide chain attached to a protein. The carbohydrate component makes up 95 percent and the protein component five percent of the molecular mass. Depending on which glycosaminoglycan and which enzyme is affected, a distinction can be made between six different main forms of mucopolysaccharidoses: These include Hurler / Scheie's disease (MPS I), Hunter's disease (MPS II), Sanfilippo's disease (MPS III), Morquio's disease ( MPS IV), Maroteaux-Lamy's disease (MPS VI), and Sly's disease (MPS VII). All types have severe and mild forms.
causes
The cause of all mucopolysaccharidoses is an increasing storage of glycosaminoglycans (GAGs) in the lysosomes of the cells. The degradation of the corresponding biopolymers is disturbed. For each individual disorder, either a certain enzyme is missing or this enzyme works incorrectly. There can be several mutations for each enzyme. The inheritance of the corresponding mutation can be autosomal recessive, autosomal dominant or x-linked recessive.
Since an enzymatic process usually involves several reaction steps, several enzymes can theoretically be mutated for the same glycosaminoglycan. The symptoms of the disorder would be the same or similar.
- At MPS I, Hurler's or Scheie's disease, the enzyme alpha-l-iduronidase is defective.
- MPS II represents the Hunters Syndrome with a defective iduronate-2-sulfatase.
- The Sanfilippo Syndrome (MPS III) can be divided into several subtypes. Several enzymes can be affected in this condition.
- Morquio's disease (MPS IV) is caused by a defective β-galactosidase.
- In Maroteaux-Lamy syndrome (MPS VI) it is N-acetyl-galactosamine-4-sulfate sulfatase.
- Sly's disease (MPS VII) is caused by defective β-glucuronidase. When the corresponding glycosaminoglycans are stored in the lysosomes, these become larger and larger.
The cells also enlarge because they need more and more space for non-degraded GAGs. This is also noticeable in the enlargement of many organs. A typical symptom is the constant enlargement of the liver and spleen. If left untreated, the storage diseases lead to death through the gradual destruction of the organs.
Symptoms, ailments and signs
The symptoms are similar for all diseases. There are severe and mild forms. However, a mild course only means that the disease is progressing more slowly. The final course is always the same. There is progressive deformation of the skeletal system, joint contractures, coarsening of facial features and enlargement of the liver and spleen.
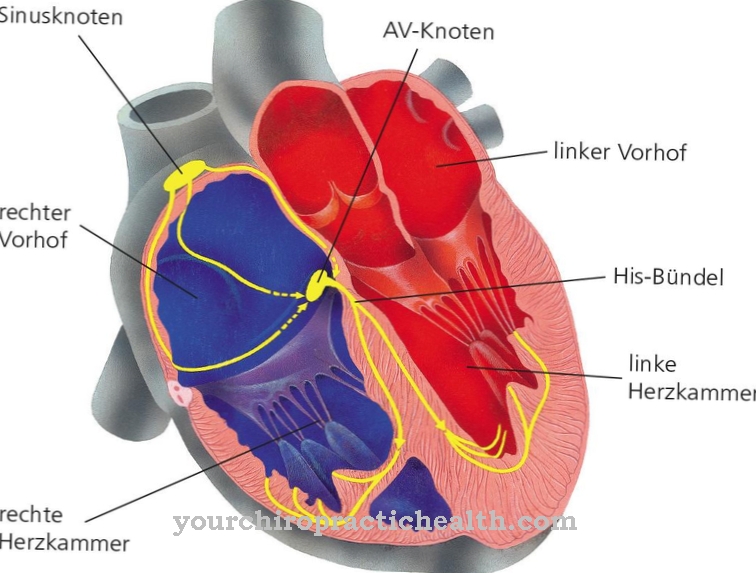
The mental and motor skills decrease in the short or long term. In the severe forms of the disorders, the clinical pictures are very similar. Umbilical and inguinal hernias, heart problems and respiratory infections occur at an early stage. Over time, the narrowing of the airways and the enlargement of the tonsils and tonsils develop into massive sleep apnea problems.
Diagnosis and course of disease
Mucopolysaccharidoses can be diagnosed by examining the urine for excreted glycosaminoglycans. In mucopolysaccharidosis, the values are always increased. The activity of the suspected defective enzyme in the leukocytes or fibroblasts can also be determined. A certain pattern of excretion of the glycosaminoglycans leads the suspicion to a corresponding enzyme, which is then examined.
Complications
Due to mucopolysaccharidosis, those affected suffer from various malformations and skeletal complaints. Deformations occur that can significantly limit the patient's everyday life. As a rule, the joints are also affected by mucopolysaccharidosis, so that the patient's movement is restricted.
Children in particular are affected and suffer from severely delayed development, so that various consequential damages can also occur in adulthood. It is not uncommon for mucopolysaccharidosis to cause problems in the heart or breathing. In the worst case, sudden cardiac death can lead to the death of the person concerned. Due to the breathing difficulties, the patients suffer from tiredness and fatigue.
The resilience of those affected also decreases enormously. It is not uncommon for breathing difficulties at night to lead to sleep problems and thus to depression. The quality of life of the patient is considerably reduced by the mucopolysaccharidosis. A causal treatment of this disease is unfortunately not possible. Those affected are therefore dependent on bone marrow donors to treat the symptoms. There are no particular complications. However, in most cases, patients are dependent on lifelong therapy.
When should you go to the doctor?
Changes and abnormalities in body structure indicate a health impairment. A doctor's visit is necessary as soon as there are permanent optical peculiarities or the person concerned has trouble deliberately optimizing their posture.Swelling of the joints, changes in facial features or enlargement of the chest should be intensively examined by a doctor so that a diagnosis can be made. A doctor is required if there are restrictions in the possibilities of movement, irregularities in everyday voluntary control and a decrease in physical and mental performance. Consult a doctor in the event of disturbances in the heart rhythm, problems with breathing or interruptions during night sleep.
Swelling in the throat, a feeling of tightness in the throat, disturbances in the act of swallowing and changes in vocalization are considered worrying. They must be examined by a doctor so that the symptoms can be alleviated. If the person concerned suffers more infections, if the ability to concentrate and pay attention decreases or if umbilical or inguinal hernias occur repeatedly, a doctor should be informed of the observations.
Sudden skin blemishes, yellowing of the skin as well as inner restlessness should be examined and treated. A doctor is needed as soon as there is pain in the body, a reduced quality of life and behavioral problems. If there is a risk of shortness of breath, an ambulance service is required. To prevent this acute condition, a doctor should be consulted as early as possible.
Therapy and Treatment
A causal therapy is not yet possible today. However, there are some approaches to future gene therapy for these diseases in research projects. Unfortunately, there are currently no tangible results in this area. However, a clinical study on gene therapy for Hurler's disease is to start in Barcelona. With some forms of mucopolysaccharidoses, bone marrow transfers have proven effective in individual cases. This affects, for example, Hunter's disease, Hurler's disease or Sanfilippo's disease.
Through this bone marrow transfer, the diseased stem cells are exchanged for healthy stem cells from a donor. This enables the organism to sufficiently restore the missing enzyme. Enzyme replacement therapy also pays off in many cases. However, this replacement therapy must be carried out for life. However, there are also cases in which promising therapies are no longer possible. However, the point here is to carry out symptomatic treatments.
You can find your medication here
➔ Medicines for painOutlook & forecast
The further development in patients with mucopolysaccharidosis must be assessed individually. This term is a collective term for various storage diseases. These are present to different degrees in each patient and their intensity is individually pronounced. If medical care is not instituted, the internal organs of all those affected are gradually destroyed over the course of their life. This results in a shortening of the average expected life.
With an early diagnosis, a personally optimized therapy can be worked out. This is tied to the health requirements and existing complaints of the patient. Long-term treatment is fundamentally necessary in order to achieve a stable improvement in health. Surgical interventions can occur, each of which is associated with different risks and side effects. If the operation goes without any further complications, an alleviation of the symptoms can usually be observed afterwards.
Nevertheless, unwanted developments and setbacks can occur in the course of life. In individual cases, only a bone marrow transplant can improve the general quality of life. Due to the overall circumstances, the patient experiences a strong emotional and mental burden. Normal everyday life is often not possible due to the symptoms. Psychological complications can occur and lead to a further deterioration of the situation.
prevention
Since the mucopolysaccharidoses are hereditary diseases, prevention is not possible. In the case of an existing illness, the success of the treatment can be ensured through timely therapy. In addition, constant monitoring of the lung and heart function is necessary. If cases of mucopolysaccharidosis have already occurred in the family, the risk of the disease can be assessed through genetic counseling if the family wishes to have children.
Aftercare
In the case of mucopolysaccharidosis, in most cases the patient has only a few options for follow-up care, so that the person affected should primarily consult a doctor at an early stage. Only with early detection and treatment of this disease can further complications be prevented, so that a doctor should be contacted as soon as the first signs and symptoms appear.
In most cases, those affected are dependent on surgical interventions, which can alleviate and limit the symptoms. However, since mucopolysaccharidosis is a genetic disease, it usually cannot be completely cured.
Therefore, the person concerned should first consult a doctor if they want to have children in order to prevent the recurrence of this disease in the children. It is often also very important to have family support during treatment. This can also prevent depression and other psychological upsets. Mucopolysaccharidosis may lead to a reduced life expectancy of the person affected, whereby the further course depends very much on the time of diagnosis.
You can do that yourself
The possibilities of self-help in the case of mucopolysaccharidosis are limited to alleviating symptoms and thus improving quality of life. Self-help groups have proven to be very helpful, as the exchange with other parents reveals valuable tips and can often alleviate fears and concerns and give a more positive view of the future.
The accompaniment to physiotherapy, occupational therapy, speech therapy and other forms of therapy, which can often be deepened in the home environment, are now an integral part of life.
In order to make life as easy as possible for yourself and the affected child, it is advisable to make the living environment accessible to the disabled as early as possible. With increasing age and weight of the child, height-adjustable care beds prove to be a great physical relief for the caregiver. Epilepsy warning devices and other technical aids enable the best possible safety even at night and relieve the parents at night so that they can sleep more relaxed.
Keeping a symptom diary can help the doctor recognize new symptoms and possibly correct the treatment of existing symptoms, since drug therapy often does not show the desired effect, but the opposite effect.
Since the disease is very demanding on relatives, they have to create small spaces for themselves to recharge their batteries. This can include cures, preventive care or, later, a vacation in the hospice.