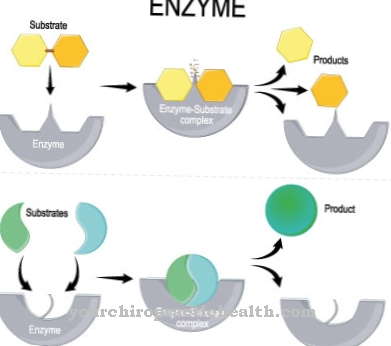
Amino acids are one of the most important substances for the human body. Without this, the metabolism cannot produce proteins, liver metabolism, growth, structure of the skin, nails and hair and the functioning of the nervous system would not be possible.
Amino acids are building blocks for the synthesis of proteins and serve as basic building blocks for gluconeogenesis, which is the metabolic pathway for the formation of glucose. Of the Amino acid metabolism thus includes all biochemical processes in the body during the composition, breakdown and conversion of amino acids.
What is the Amino Acid Metabolism?

The human body can produce some amino acids itself and gets others from food. The processes involved are complex.
Enzymes and coenzymes are used, which in the event of a defect or insufficient formation of such enzymes lead to disorders of the amino acid metabolism, which in turn leads to serious diseases.
Individual amino acids also form precursors of hormones and neurotransmitters or are used in the composition of nitrogenous compounds.
Function & task
Basically, amino acids are divided into essential and non-essential. The formation and breakdown of amino acids is part of the amino acid metabolism.
The human organism cannot produce essential amino acids by itself, since in the course of evolution the appropriate enzymes were gradually lost and the synthesis is rather cumbersome. They are ingested through food and are called leucine, isoleucine, leucine, methionine, lysine, phenylalanine, tryptophan, threonine and valine. In the case of amino acid metabolism diseases, other amino acids are also essential, e.g. B. in phenylketonuria tyrosine.
The body can produce non-essential amino acids by itself. Such are e.g. B. aspartic and glutamic acid, alanine or serine. The formation takes place in the liver and kidneys, for the processes in the liver the body needs alpha-keto acids which are made available by beta-oxidation from fatty acids.
Alpha-keto acids serve to transfer amino groups through amino acids. The body gains a derivative from vitamin B6, which is called the coenzyme pyridoxal phosphate and which plays an important role in the transmission. In amino acid metabolism, the coenzyme is also responsible for dehydration, transamination and decaroxidation into biogenic amines.
Dehydrogenation is the process of separating hydrogen from molecules, meaning organic compounds that have been oxidized. This takes place in an endothermic reaction, but it can also be converted into an exothermic reaction by oxygen, which then forms water.
During transamination, the alpha-amino group of an amino acid is shifted to an alpha-keto acid, creating a new alpha-keto acid and amino acid. The enzymes required for this are called transaminases. Acceptors are ketoglutarate, oxaloacetate and pyruvic acid. The process that takes place in this way is always reversible.
Decarboxylation is the splitting off of a carboxyl group from a molecule and the associated formation of carbon dioxide. Biogenic amines are combined in this way, which in turn are basic molecules that carry amino groups.
In the case of non-essential amino acids, the aldehyde group of the coenzyme pyridoxal phosphate and the amino group of the acid form a Schiff's base with the elimination of water. The Schiff base is thus stabilized by a cationic group inside the enzyme. In the pyridine ring of the coenzyme there is still an electrophilic effect of nitrogen and the formation of a ketime information that is not stable and is converted. This can be done either via transamination or deamination.
The latter involves splitting off an amino group from an amino acid and is reused as ammonia. This is e.g. B. important for the pH values of the blood, but is recognized by the brain as toxic at low concentrations and therefore converted in the liver to urea, which is then excreted.
You can find your medication here
➔ Medicines for muscle weaknessIllnesses & ailments
If defects occur in the amino acid metabolism, serious damage to the organism and associated diseases arise. Most are innate gene effects that are transmitted through heredity. Many of them are rather rare and their manifestations depend on whether enzymes are missing or just impaired in their function.
Several processes take place in the amino acid metabolism, which in turn require different enzymes. The disturbances can also vary. The diagnosis of such diseases is all the more difficult. Symptoms are not always known and are also difficult to identify.
Most defects manifest themselves in the fact that intermediate products can no longer be converted into amino acids. The preliminary stages to this process can also be blocked. The associated consequences are an accumulation of such intermediate products, whereby the functions can no longer take place due to the lack of amino acids. This soon results in tissue and organ damage or failure symptoms. Under these conditions the amino acid transport is disturbed. Amino acids are no longer excreted in the urine, but transported back into the organism.
Diseases resulting therefrom are e.g. B. albinism, alkaptonuria and phenylketonuria. In all three manifestations, the tyrosine metabolism is disturbed. For this reason, in albinism, the human body can no longer produce the skin pigment melanin. The skin, eyes and hair are therefore very light, pink to white.
Tyrosine is made from the amino acid phenylalanine, which is obtained from food. If this is missing, the thyroid hormone thyroxine or messenger substances such as catecholamines can no longer be formed in addition to melanin. In alkaptonnuria, the enzyme homogentisine oxygenase is missing. This converts homogeneous thisic acid into maleyl acetic acid. As an alkapton, this is excreted in the urine or stored in the body. This causes inflammation caused by crystal or calcium deposits in the joints, as well as kidney stones or cardiac dysfunction. One possible diagnosis is the addition of bases, which causes the urine to turn black.
Phenylketonuria is the most common amino acid metabolism disorder. It is triggered by a defect in the enzyme phenylalanine hydroxylase, which is responsible for converting phenylalanine into tyrosine. If this does not take place, phenylalanine is stored in the tissue and blood and begins to damage the brain. This influences and delays the entire physical development. Seizures and mental retardation can occur.
For this defect there is a diet enriched with special amino acids which, when used in the first two months of a child's life, leads to normal development. The diet must be followed until puberty.













.jpg)