The Hemoglobin synthesis is composed of the heme synthesis and the globin synthesis. Finally, the prosthetic heme group, each with four globins, is linked to the iron-containing protein complex hemoglobin. Disturbances in both heme synthesis and globin synthesis can lead to serious health problems.
What is Hemoglobin Synthesis?

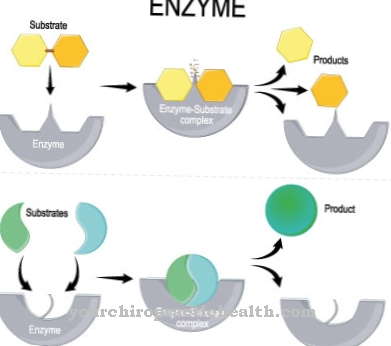
In order to understand hemoglobin synthesis, knowledge of the structure of hemoglobin is first necessary. Hemoglobin is an iron-containing protein complex, which consists of four subunits of globin, each with a prosthetic heme group.
In human adult hemoglobin there are two identical alpha globins as well as two identical beta globins as subunits. Each of these subunits is bound to a prosthetic heme group, which consists of a porphyrin iron (II) complex. Thus a hemoglobin complex contains four heme groups.
Depending on the chemical environment, each heme group can bind an oxygen molecule to the ferrous ion in a complex manner. Depending on how many heme groups are loaded with oxygen, one speaks of oxyhemoglobin (high in oxygen) or deoxyhemoglobin (low in oxygen).
The ferrous ion is located in the middle of the porphyrin ring. At the side there is a complex bond to the histidine residue of globin. On the other hand, depending on the energy state of the iron ion, an oxygen molecule can be bound in a complex. The energy state is influenced by external physical and chemical conditions due to changes in the conformation of the globin.
Function & task
The last step in hemoglobin synthesis consists in assembling the prosthetic heme group with the four globin units to form an iron-containing protein complex. The individual components are formed by independent biosynthetic pathways.
The starting materials for the porphyrin ring of the heme group are the amino acids glycine and succinyl-CoA. Succinyl-CoA is composed of coenzyme A and succinic acid. Succinic acid is an intermediate product in the breakdown of energy-rich ketone bodies as part of the energy metabolism. With the help of the enzyme delta-aminolevulinic acid synthase, delta-aminolevulinic acid is synthesized from succinyl-CoA and glycine. Two molecules of delta-aminolevulinic acid condense with elimination of one molecule of water to form the pyrrole derivative porphobilinogen. With the elimination of ammonia and with the help of the enzyme uroporphyrinogen-I synthetase, four molecules of porphobiliogen react to form hydroxymethylbilane. This is transformed into uroporphyrinogen III with ring formation.
Protoporphyrin is produced through enzymatic decarboxylation and dehydration in the mitochondria. With the enzyme ferrochelatase, an iron (II) ion is incorporated into this molecule with the formation of heme. In the cytosol of the cell, the heme is linked with the protein globin to form the iron-containing protein complex hemoglobin.
The synthesis of the individual globins takes place via normal protein biosynthesis. As already mentioned, the adult hemoglobin complex contains two identical subunits of alpha and beta globins. Due to its complex structure, the finished hemoglobin has developed the ability to transport oxygen and supply it to all cells of the organism.
However, the binding of the central iron to oxygen is not very tight and can be influenced very easily by external chemical and physical factors. This enables hemoglobin to both absorb and release oxygen quickly. The oxygen content of the hemoglobin depends, among other things, on the factors pH, carbon dioxide or oxygen partial pressure or temperature. These influencing variables change, for example, the conformity of the globins, so that the oxygen bond can be strengthened or weakened by slight changes in the energetic and steric conditions.
With a low pH value and a high carbon dioxide partial pressure, the oxygen bond to the iron (II) ion is weakened and thus the release of oxygen is favored. Exactly under these conditions stronger metabolic turnover takes place, which also have an increased oxygen demand. The oxygen transport system is therefore optimally coordinated with the physical needs via the hemoglobin function.
Illnesses & ailments
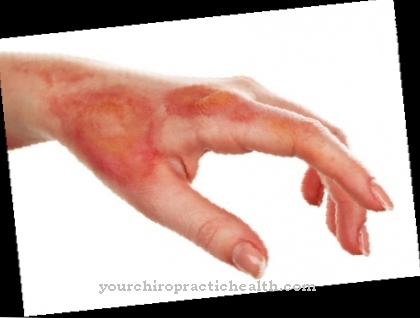
Disturbances in hemoglobin synthesis can lead to various diseases. There are a number of genetic diseases that are based on the disruption of the synthesis of heme. In the process, heme precursors accumulate in the body, which among other things lead to extreme sensitivity to light. In these so-called porphyrias, porphyrins are stored in the blood vessels or even the liver. When exposed to light, some forms of porphyria store more radiation energy. When the energy is released, oxygen radicals are created which attack and destroy the exposed tissue. This leads to severe itching and burning pain.
There are seven forms of porphyry. The construction of the heme is an eight-step process in which seven enzymes are involved. If an enzyme is only functioning inadequately, the respective precursor is stored at this point in the heme synthesis. Based on the symptoms, the porphyrias are divided into two main groups. The so-called cutaneous porphyrias are characterized by the painful sensitivity of the skin to light. In hepatic porphyrias, liver involvement predominates with severe abdominal pain, nausea and vomiting. In many cases, however, there is an overlap between the two symptom complexes.
Porphyrias often show an intermittent course with acute attacks. Depending on the type of porphyria, these manifest themselves in suddenly painful skin reactions, colic-like abdominal pain, nausea / vomiting, red coloration of the urine, seizures, neurological deficits or even psychoses.
Other disorders of hemoglobin synthesis relate to the faulty synthesis of globin molecules through mutations in the corresponding genes. Examples are the so-called sickle cell anemia or thalassemia. In sickle cell anemia, the protein of the beta globin subunit is genetically modified. In position six of this protein, the amino acid glutamic acid has been replaced by valine. If there is a lack of oxygen, the hemoglobin concerned becomes sickle-shaped, clumps together and clogs small blood vessels. This results in life-threatening circulatory disorders. Thalassemias are a group of different hemoglobin malformations that lead to reduced globin chain formation of alpha or beta globin.Severe anemia is the most important symptom.













.jpg)