As Homocystinuria is a rare, genetically determined metabolic disease that can be traced back to an enzyme deficiency and is characterized by an increased concentration of homocysteine in the blood. Homocystinuria can usually be treated well with early and consistent therapy.
What is homocystinuria?

© Anastasia Okhrimenko - stock.adobe.com
As Homocystinuria is a rare, genetically caused amino acid metabolism disease, which is due to defects in various enzymes involved in methionine metabolism (essential amino acid).
Homocysteine and homocystine are degradation or intermediate products of this metabolic process and are immediately further metabolized in healthy people. Due to the presence of defective enzymes, this is only possible to a limited extent in those affected by homocystinuria, so that the concentration of homocysteine in the blood and homocystine in the urine is increased.
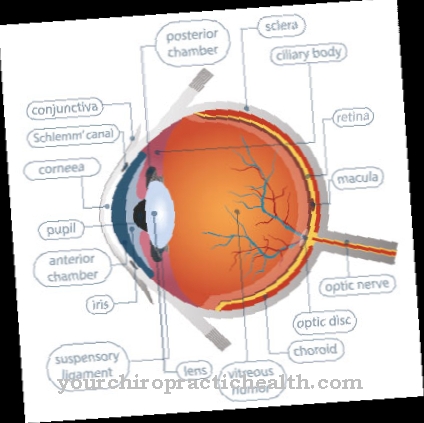
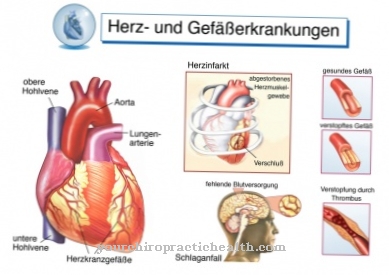
The increased concentration of these amino acids, which are considered toxic, can damage various organ systems. Eye diseases (lens dislocation, myopia, glaucoma), skeletal changes (osteoporosis, marfanoid long limbs), damage to the central nervous system (mental and physical retardation, spasms, cerebrovascular disorders) and the vascular system (thromboembolism, vascular occlusion) are characteristic sequelae of homocystinuria.
causes
Homocystinuria is due to an autosomal recessive inherited genetic defect, which results in a deficiency of various enzymes involved in methionine metabolism. Depending on the specifically affected enzyme and sub-process of methionine metabolism, three forms of homocystinuria are distinguished.
With the more frequent Type I. In homocystinuria, there is a deficiency in the enzyme cystathione beta synthase (CBS), which disrupts the synthesis of cysteine from methionine. As a result, homocysteine builds up in the blood (hyperhomocysteinemia) and homocystine builds up in the urine (homocystinuria).
Type II Homocystinuria is characterized by a lack of 5,10-methylenetetrahydrofolate reductase (MTHFR), which regulates methionine synthesis from homocysteine. This metabolic sub-process is accordingly disturbed in type II sufferers and, in addition to the enrichment of the serum with homocysteine, can also lead to a methionine deficiency.
Type III homocystinuria is characterized by a cobalamin deficiency (coenzyme vitamin B12). Cobalamin is also involved in the methionine synthesis from homocysteine, so that a deficiency can also cause an increased homocysteine concentration in the blood and a methionine deficiency.
Symptoms, ailments & signs
Homocystinuria can occur in different forms. The symptoms are diverse and differ depending on the stage of life. Before the age of two, signs of the disease only appear in particularly rare cases. Apart from characteristic laboratory findings, newborns with homocystinuria are normal.
Typically, the homocysteine level in the blood is significantly increased. This damages the blood vessels, which in the long term can lead to vascular calcification (arteriosclerosis) and the associated embolism and thrombosis. As a result, the life expectancy of those affected is significantly limited. The most noticeable symptom of childhood metabolic disorder is a prolapse of the lens of the eye.
This is often associated with myopia. The earlier the first signs of the disease appear, the higher the risk of psychomotor retardation (intellectual disability), which is irreversible. Most of those affected already have osteoporosis in childhood. As a result, the spine flattens out and gradually deforms.
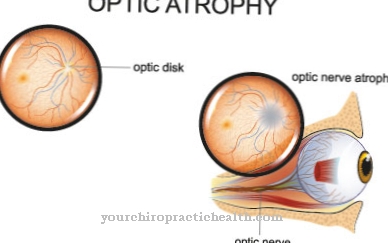
The high homocysteine level often leads to tall stature and symptoms that can be similar to Marfan's syndrome. These include the presence of a chicken breast and funnel breast, a displaced eye lens (lens dislocation or lens ectopy), glaucoma (glaucoma), retinal detachment and spider fingers (arachnodactyly).
Diagnosis & course
To diagnose Homocystinuria different laboratory analyzes are used. If a urine analysis (e.g. cyanide-nitroprusside test) reveals an increased homocystine concentration and / or a decreased methionine concentration (types II and III), this can indicate homocystinuria.
A blood analysis can be used to determine the serum homocysteine concentration and diagnose hyperhomocysteinemia associated with homocystinuria. The diagnosis is confirmed by culturing cells from a connective or liver tissue sample, whereby the underlying genetic defect can be detected directly.
The course of homocystinuria can vary from person to person in terms of symptoms and secondary diseases. With an early diagnosis and an early start of therapy, homocystinuria usually has a favorable course and a good prognosis.
Complications
Homocystinuria primarily leads to severe psychological complaints that can have an extremely negative impact on the patient's life and everyday life. In most cases, a strong personality disorder occurs, which is accompanied by behavioral disorders. These disorders can lead to severe complications, especially in children.
Usually the patient is affected by social exclusion and withdraws more and more from life. It is not uncommon for this to lead to depression. Furthermore, there are discomforts in the eyes, so that, for example, a glaucoma or myopia can develop. Various diseases of the blood vessels also occur much earlier and can lead to restrictions in movement.
Treatment itself, as a rule, does not lead to particular complications and is carried out with the help of medication. The disease progresses positively relatively quickly. In most cases, there are no further symptoms even after the treatment. Early treatment does not reduce life expectancy. Psychological complaints can be treated supportively by a psychologist.
When should you go to the doctor?
If symptoms such as behavioral disorders, thrombosis, or delays in development occur, a doctor should definitely be seen. Signs of osteoporosis or arteriosclerosis should also be clarified at an early stage. A doctor must determine the cause of the symptoms and, if necessary, initiate treatment. Therefore, the symptoms mentioned should be clarified quickly. People with a genetic defect are particularly prone to developing homocystinuria. Those affected should consult their family doctor closely and inform them of any unusual symptoms.
Basically, complaints must be clarified that persist for more than a few days or that increase in intensity over a longer period of time. The characteristic signs of homocystinuria usually develop insidiously and are often only recognized when irreversible diseases have already set in. It is all the more important to recognize the early symptoms and have them treated. People who notice physical or mental changes in themselves or others that may be related to metabolic disorders should speak to their family doctor as soon as possible. Other contact persons are specialists in internal medicine or a specialist clinic for hereditary diseases.
Doctors & therapists in your area
Treatment & Therapy
The therapy one Homocystinuria depends on the underlying disease type or enzyme defect and aims to reduce and eliminate the increased concentration of toxic homocysteine. Type I homocystinuria is treated with pyridoxine (vitamin B6) when there is residual activity of the defective enzyme.
The substance increases the enzyme activity and lowers the homocysteine concentration in the blood. About 50 percent of those affected by this type respond very well to oral therapy with high-dose vitamin B6. In addition, a diet low in methionine and high in cystine is recommended to support therapy.
If there is residual enzyme activity in type II and III homocystinuria, in which methionine synthesis from homocysteine is disturbed, attempts are made to limit the impairment with cobalamin preparations (vitamin B12). For both forms of homocystinuria, a methionine-rich diet is indicated.
In addition, folic acid, which also has a positive effect on the activity of the defective 5,10-methylene tetrahydrofolate reductase, as well as methionine and betaine are used therapeutically for type II. Anticoagulant drugs (acetylsalicylic acid) are used to help prevent vascular diseases such as thrombosis and embolism.
Outlook & forecast
With early diagnosis and intensive therapy, the prognosis for homocystinuria is usually favorable. The disease is not curable because it is a genetic defect. However, as part of the therapy, it is possible to permanently reduce the concentration of homocysteine and methionine, which significantly reduces the likelihood of developmental disorders and complications.
The degree of severity of homocystinuria can be varied. There are forms of the disease that are initially inconspicuous and are otherwise mild. However, even here from the age of 20 or 30 there is a greater risk of developing arteriosclerosis, thrombosis, embolism, heart attacks and strokes.
However, if the homocysteine concentration is already very high in infancy, there is a great risk of physical and mental development disorders in the child without intensive treatment. A mental handicap can already become apparent in the first two years of life. The affected children often also suffer from osteoporosis. Up to 70 percent of untreated children develop eye problems, which are most clearly expressed in a prolapse of the eye lens. Other consequences for the eyes are glaucoma, extreme myopia, retinal detachment and blindness. If severe forms of the disease are treated too late or not at all, thromboses and embolisms will develop in 30 percent of all patients under the age of 20.
prevention
There Homocystinuria is a genetic disease, it cannot be prevented. However, if treatment is started early, the complications of homocystinuria can be prevented or limited. In addition, those affected have the option of having their unborn child tested for homocystinuria as part of a prenatal diagnosis (amniotic fluid analysis). Siblings of those affected are also recommended to be checked for homocystinuria.
Aftercare
Depending on the type of enzyme defect that is the cause of the metabolic disorder, a whole range of measures are useful and necessary as part of the follow-up care for homocystinuria. With homohystinuria type I, the patient must adhere to a diet rich in vitamins in addition to the medical administration of vitamin B6. Vitamin B6 increases the activity of the defective enzyme and consequently leads to a lower concentration of homocysteine in the blood.
The diet must be adhered to permanently so that this effect is maintained. The same applies to the consumption of foods rich in cystine and low in methionine. In type II homocystinuria, the diet must also be continued in order to achieve long-term effects. In addition, regular follow-up checks apply during follow-up. The doctor must check the activity of the affected enzymes and adjust the therapy if necessary.
Since homocystinuria is usually not a serious illness, medical checks every three to six months are sufficient. In the case of severe disorders, a specialist should be consulted monthly after the actual therapy has been completed. In addition, attention must be paid to unusual symptoms, as the metabolic disorder can cause other diseases in the long term that must be treated.
You can do that yourself
Depending on the type of enzyme defect underlying homocystinuria and the therapy the doctor is using, the patient can do a few things himself to alleviate the symptoms.
First of all, a diet rich in cystine is important. The victim should mainly consume rice, nuts, soybeans and oat products. The active ingredient is also found in watermelons, sunflower seeds and green tea. The doctor will also prescribe vitamin B12 supplements to limit the impairment. The person affected can support these measures by working with a nutritionist to create a methionine-rich nutrition plan and implement it consistently. Foods high in protein such as eggs or meat should be avoided. Low-protein foods are allowed, including fruit, vegetables and low-protein pasta, bread or flour from specialist shops. This diet should also be supported with various B vitamins and folic acid.
After the disease has resolved, the patient should have additional check-ups. Homocystinuria is a lifelong disease that requires regular evaluation in a specialized treatment center. Close control enables metabolic problems to be identified and treated at an early stage before complications arise.











.jpg)