Of the Medium chain acyl CoA dehydrogenase deficiency (MACD deficiency) is a genetic metabolic disorder in which medium-chain fatty acids are only insufficiently broken down. Under certain conditions, dangerous metabolic imbalances can occur, which can be fatal under certain circumstances. If therapy is started early, the disease can be easily controlled.
What is Medium Chain Acyl CoA Dehydrogenase Deficiency?

© royaltystockphoto - stock.adobe.com
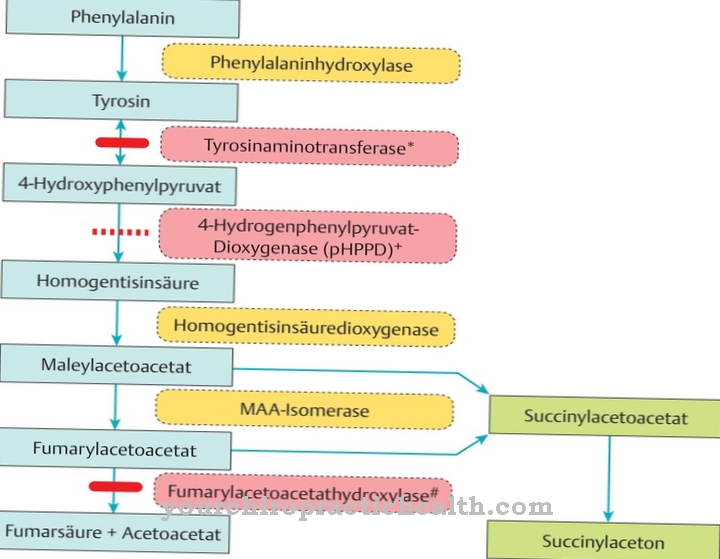
At the Medium chain acyl CoA dehydrogenase deficiency the breakdown of medium-chain fatty acids is disturbed due to genetic causes. These can no longer be used sufficiently to generate energy. Therefore, the body is forced to break down carbohydrates and amino acids to a greater extent.
When the energy requirement increases due to higher physical stress, growth processes or infections, the body's carbohydrate reserves and the body's own proteins are used to a greater extent for energy production. The same applies to longer periods of no food. Normally, under these conditions, the breakdown of fatty acids is forced through beta-oxidation.
However, this is with one MACD deficiency not sufficiently possible. The resulting increased breakdown of glucose leads to dangerous hypoglycaemia. Due to the forced breakdown of amino acids, the ammonia level in the blood increases at the same time. Carnitine, which is important for energy metabolism, is reduced because it forms compounds with the accumulating medium-chain fatty acids.
The compounds represent intermediate products of fatty acid breakdown. Since the further breakdown of the medium-chain fatty acids is blocked by a defective enzyme in the acyl-hydrogenase complex, a secondary carnitine deficiency develops. A large deviation from the corresponding values can lead to life-threatening complications. If left untreated, about 25 percent of the metabolic imbalances caused by this disorder are fatal.
Overall, the medium-chain acyl-CoA dehydrogenase deficiency is one of the most common metabolic diseases and is part of the examinations as part of a newborn screening. A timely determination of the responsible genetic defect is necessary for an effective therapy. This disease can be easily treated with suitable therapeutic measures.
causes
The cause of the medium chain acyl-CoA dehydrogenase deficiency is to be found in a malfunction of the MCAD enzyme. Due to an autosomal recessive mutation of the MCAD gene, which is located on chromosome 1, the enzyme's folding properties are impaired. The result is a misfolded enzyme, which is eliminated and broken down as part of a protein quality control.
As a result, there is not enough medium chain acyl CoA dehydrogenase. Medium-chain fatty acids are then only poorly broken down. As mentioned earlier, an autosomal recessive mutation in the MCAD gene is responsible for this disease. In the case of autosomal recessive inheritance, the affected patient has two defective genes, one copy of which was transferred from both parents.
People with only one mutated gene do not get the disease. Therefore, the disease is not passed on from generation to generation. If both parents are heterozygous for a defective gene each, the offspring has a 25 percent chance of developing MCAD deficiency.
Symptoms, ailments & signs
Medium chain acyl-CoA dehydrogenase deficiency is characterized by a tendency to hypoglycaemia, convulsions and comatose states. The first symptoms usually appear between the second month of life and the fourth year of life. They are triggered by infections or prolonged periods of abstinence from eating and manifest themselves as diarrhea, vomiting, impaired consciousness and seizures.
The liver may be enlarged. With every metabolic derangement, the blood sugar values are lowered, the ammonia values in the blood are increased and the carnitine level is lowered. The greater the deviations, the greater the risk of falling into a life-threatening coma. However, there are also forms of MCAD deficiency with much milder symptoms.
In some cases there are no symptoms at all. Here only laboratory tests indicate a corresponding genetic defect. Even in the more severely ill patients, there are always symptom-free phases between the metabolic imbalances.
Diagnosis & course of disease
A medium chain acyl CoA dehydrogenase deficiency can be detected in the context of a newborn screening using tandem mass spectroscopy. In this method, fragments of acyl carnitines are determined. The concentration of octanoylcarnitine is the key parameter here. If the levels are elevated, a DNA analysis can help confirm the diagnosis.
In the acute phase of the disease, hypoglycemia, elevated ammonia levels and elevated levels of medium-chain fatty acids in the form of dicarboxylic acids can be detected in the urine. There is also hyperuricaemia (increased uric acid levels in the urine) and signs of liver dysfunction. Sometimes urine myoglobin levels are also increased.
Complications
The medium chain acyl-CoA dehydrogenase deficiency can, in the worst case, be fatal for the patient. In order to avoid serious complications and consequential damage, early treatment of this disease must take place. In most cases, those affected suffer from severe cramps, which are also associated with pain.
The medium chain acyl-CoA dehydrogenase deficiency can also lead to a coma or loss of consciousness, so that the everyday life of the person affected is significantly restricted by the complaint. Disturbances in consciousness occur and those affected suffer from diarrhea and vomiting. In many cases, the liver also enlarges, which is also associated with pain.
If the person concerned is in a coma, this condition can also have a negative effect on the psyche of relatives or parents and children and lead to depression or other psychological disorders in these people. Usually, medium chain acyl-CoA dehydrogenase deficiency can be treated relatively well if it is diagnosed early.
There are no particular complications. Only with a late diagnosis and treatment can various complications arise, so that the person affected is dependent on an operation, for example.
When should you go to the doctor?
Food cravings and related symptoms such as nausea and vomiting are signs of a health problem. A doctor is required if there are disorders of concentration, malaise or a decrease in performance level. In the event of pain, cramps and a seizure disorder, a doctor should be consulted immediately. Loss of drive, loss of appetite or an increased susceptibility to infections should be examined and treated. Since the disease shows its first symptoms between the ages of six months and four years, small children in particular are at risk of developing the disease. If the affected person shows a noticeable change in their weight, if there is indifference or behavioral problems, a doctor should be consulted.
Increased vigilance is necessary in the event of disorders of consciousness. As the risk of an accident or injury increases due to the reduced awareness of the person concerned, action is required. If a comatose state occurs or if there is a loss of consciousness, an ambulance service should be alerted. At the same time, first aid measures are required until it arrives. Swelling of the upper body, tiredness, exhaustion as well as inner restlessness and increased irritability should be presented to a doctor. The disease is characterized by the gradual regression of all symptoms. Despite the symptom-free period, it is advisable to see a doctor as soon as signs of a health impairment appear repeatedly.
Treatment & Therapy
If the diagnosis is made early, the prognosis for the disease is very good. Then prophylactic measures can be initiated in good time to prevent metabolic crises even in difficult and stressful situations. It is important to avoid long periods of abstinence from food and infections.
Food leave should not last longer than two hours. Knowledge of this disease is also necessary in preparation for any operations. Many operations that are not absolutely necessary should therefore not be carried out. If an operation cannot be avoided, artificial nutrition with glucose solution is essential to prevent metabolic imbalance.
Even an acute metabolic imbalance can only be overcome by an infusion with glucose solution. Since there is no carnitine, oral administration of carnitine also makes sense. In addition, the administration of riboflavin can lead to an improvement in fat metabolism, because it is a coenzyme in the breakdown of fatty acids.
Outlook & forecast
In the presence of a medium chain acyl CoA dehydrogenase deficiency, the prognosis is positive if the diagnosis is made as early as possible. However, under certain conditions this congenital metabolic defect can lead to serious metabolic imbalances with fatal consequences.
The problem is that a medium-chain acyl-CoA dehydrogenase deficiency is variable in the clinical picture. Therefore, misdiagnosis cannot be ruled out. The symptoms of medium chain acyl-CoA dehydrogenase deficiency are already noticeable in newborns. In order to improve the prospects for those affected, metabolism screening is usually carried out on newborns today.
Living conditions are good as long as the person concerned avoids long periods of being sober or starving. He should have a low-fat diet that contains sufficient amounts of carbohydrates. In addition, a carnitine deficiency must be taken care of. In order to prevent an impending metabolic imbalance, the administration of glucose-electrolyte infusions can also be considered.
Emergencies and impending metabolic imbalances represent a high risk. Therefore, those affected by medium-chain acyl-CoA dehydrogenase deficiency should immediately be referred to inpatient treatment. Those affected by MCAD carry an emergency ID card with them, which identifies them as patients with a medium chain acyl-CoA dehydrogenase deficiency.
The outlook for people with medium chain acyl-CoA dehydrogenase deficiency is worse if they suffer from infections or if they fast. Sobriety before an operation also carries great risks. Metabolic imbalances can lead to hypoketotic hypoglycaemia or metabolic acidosis. If neurological symptoms such as hypotension or lethargy occur, unconsciousness or death can occur.
prevention
Because medium-chain acyl-CoA dehydrogenase deficiency is genetic, there is no recommendation for its prevention. If there is more than one family member, genetic counseling is useful if there is a desire to have children. The defective genes can be detected by DNA examinations.
If both parents have a mutated gene, the offspring have a 25 percent risk of medium-chain acyl-CoA dehydrogenase deficiency. If an MCAD deficiency is found in the newborn screening, a comprehensive medical check-up is necessary to prevent infection. The diet must be adjusted to a high-carbohydrate and low-fat diet.
Aftercare
Since peroxisomal disease is based on a genetic defect, therapy can only alleviate a few symptoms without being able to cure the disease completely. For this reason there is no actual aftercare treatment in this sense, but intensive symptom treatment to alleviate some ailments.
In general, those affected and their relatives are advised to lead a healthy lifestyle with a balanced diet to support the immune system. He will also suggest certain relaxation and mental techniques for stabilization and advise on as many leisure activities as possible. Mental equilibrium makes it easier to deal with the disease and can sometimes promote recovery. If the parents who have already been affected wish to have children again, a detailed genetic examination is recommended to determine the likelihood of another sick child in advance.
You can do that yourself
Medium chain acyl CoA dehydrogenase deficiency is a metabolic disease that can be treated well if it is detected early. An important self-help measure is to regulate your metabolism through diet and sufficient exercise. People who regularly take medication or engage in activities that negatively affect the metabolism should speak to their family doctor. Appropriate therapy can optimize the metabolism and thus the MCADD can also be treated well.
In addition to medical treatment, alternative therapy is also possible. Natural remedies such as valerian or sage can have a good influence on the healing process due to their pain-relieving effect and other positive effects. In order to minimize any risks and side effects, the use of such preparations should first be discussed with the responsible doctor.
Parents whose child died of MCADD should seek trauma therapy. In the event of a new pregnancy, screening should be carried out at an early stage so that a possible medium chain acyl-CoA dehydrogenase deficiency can be determined and sudden infant death can be averted. The affected children require constant observation so that the medical emergency service or the ambulance service can be contacted immediately in the event of any complications.
.jpg)
.jpg)











.jpg)