Gaucher's disease is one of the most common lipid storage diseases and can be traced back to a genetic deficiency of the enzyme glucocerebrosidase. In a large number of cases, the disease can be treated as part of enzyme replacement therapy, which causes the symptoms characteristic of Gaucher's disease to regress.
What is Gaucher's Disease?
.jpg)
© designua - stock.adobe.com
As Gaucher disease (Gaucher syndrome) is a genetic disorder of lipid metabolism caused by a deficiency in the enzyme glucocerebrosidase. As a result of the breakdown caused by this breakdown, more glucocerebroside is accumulated in the organism, especially in the reticular cells (fibroblasts in the connective tissue), which leads to an enlargement of the affected organs.
Gaucher's disease is differentiated into three forms, which differ in terms of symptoms and course. The visceral or non-neuronopathic form mainly characterizes organic impairments such as liver and spleen enlargement (hepatosplenomegaly), anemia (anemia), bone and joint disorders and coagulation disorders.
In the presence of acute neuronopathic Gaucher's disease, those affected also show damage to the nervous system. This form of the disease has a severe and highly progressive course and leads to death in the first few years of life. The chronic neuronopathic form is characterized by a slightly progressive course with a manifestation in later years of life.
causes
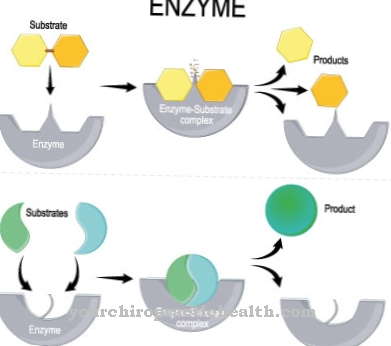
Gaucher's disease is an autosomal recessive inherited deficiency of the enzyme glucocerebrosidase, which is due to mutative changes in the genetic make-up. Glucocerebrosidase is an enzyme that helps break down glucocerebroside, a component of fat that is produced when used blood cells are broken down.
The cells of people with Gaucher's disease are not able to produce this enzyme in sufficient quantities or produce glucocerebrosidase in a reduced quality. As a result, there is an accumulation of glucocerebroside in the macrophages (phagocytes).
The macrophages collect with the undigested glucocerebroside mainly in the spleen, liver and bone marrow and cause the organ enlargement typical of Gaucher disease, which restricts the function of the affected organs.
Symptoms, ailments & signs
Gaucher's disease can come in three different forms, each with different symptoms. The mildest form of the disease is type I with a non-neuronopathic course. Type II characterizes the acute neuronopathic form, while type III of the disease is the chronic neuronopathic form. In Gaucher's disease type I, the first symptoms usually only appear in adulthood. In contrast to the other forms of the disease, there are no neurological complaints here.
However, the internal organs are affected. The spleen in particular enlarges and causes such complaints as enlarged abdomen, upper abdominal discomfort and a constant feeling of satiety. At the same time, the blood cells are broken down more quickly and blood formation in the bone marrow is hindered.
This leads to increasing anemia, which manifests itself as tiredness and exhaustion. The immune system is weakened by the lack of white blood cells. Blood clotting is also reduced because too few platelets are produced. In addition, there is deformation of the bones, increased bone fractures and frequent infections.
Bone pain, chronic joint pain and circulatory disorders are also possible symptoms in the non-neuronopathic form. In type II Gauchers syndrome, the disease begins in infants with severe symptoms that can be traced back to nerve breakdown processes.
Increasing brain damage leads to difficulty swallowing and severe seizures. Death occurs within two years. In the chronic neuronopathic form, slow nerve breakdown processes take place with progressive mental breakdown, movement disorders, behavioral problems and increasing seizures.
Diagnosis & course
The diagnosis of Gaucher's disease is based on the symptoms characteristic of the disease, such as enlargement of the spleen and liver (hepatosplenomegaly), bone and joint pain, spontaneous fractures, decreased muscle tone, anemia, signs of fatigue, seizures and changes in the fundus (white spots).
The diagnosis is confirmed by a blood test and an enzyme test in which the enzyme activity in the leukocytes or fibroblasts is determined.If an increase in phosphatase and a reduced glucocerebrosidase concentration are found in the blood and Gaucher cells can be detected in the bone marrow, the diagnosis is considered confirmed.
If therapy is started in good time, the visceral and chronic neuronopathic form of Gaucher disease can be expected to slow down the progressive course of the disease and to relieve the symptoms in most cases. The acute neuronopathic form, on the other hand, has a strongly progressive course and those affected by this form of Gaucher's disease often die in early childhood.
Complications
First and foremost, Gaucher's disease leads to a strong enlargement of the spleen. This can also lead to pain, which can significantly reduce the patient's quality of life. In most cases, patients also experience an enlarged abdomen and pain in the abdomen.
In addition to the pain, there is also a loss of appetite, which can further lead to malnutrition or a lack of nutrients. The anemia also makes the patient feel exhausted and tired. The quality of life is significantly reduced by Gaucher's disease. Pain in the joints or seizures can also occur and make everyday life difficult for the person concerned.
It is not uncommon for very young children in particular to die of the symptoms of this disease, which can lead to psychological complaints or depression in the parents and relatives of the patients. Treatment of this disease can take place with the help of infusions and other drugs.
There are usually no complications. However, it is not possible to completely limit Gaucher's disease, so that in most cases patients are dependent on lifelong therapy. Whether there is a reduction in life expectancy depends heavily on the treatment and the severity of the disease.
When should you go to the doctor?
In the case of hereditary fat storage disease Gaucher disease, those affected must expect that this seldom occurring enzyme defect will be misdiagnosed several times. Currently only about 2,000 patients are registered with it in Germany. Therefore, every visit to the doctor that becomes necessary due to this genetic defect is initially a problematic one. The fact that the symptoms are different for each person is difficult.
People who are affected by Gaucher's disease often fall ill as a child. Some people have hardly any symptoms. As a result, you do not visit a doctor. In the event of a precarious complaint due to the enzyme defect, a specialist should be consulted. There are enough tests and lists of symptoms on the Internet to make a comment to the doctor if you suspect Gaucher disease. This hint is probably the trigger for a blood test.
There is no cure for Gaucher's disease. However, it can be easily treated with enzyme replacement therapy or substrate reduction therapy. Therefore, after the diagnosis, those affected have to go to a designated Gaucher center several times to initiate appropriate treatment. In addition, regular check-ups are necessary every three to six months.
The necessary examinations should be performed in a Gaucher clinical center. Enzyme replacement therapy is also started here as infusion therapy. The family doctor can take over further treatment later.
Treatment & Therapy
At Gaucher's disease There are basically two forms of therapy available: enzyme replacement therapy (enzyme replacement therapy - EET) and substrate reduction therapy. Since Gaucher's disease is caused by an enzyme deficiency, treatment focuses on eliminating this deficiency with enzyme replacement therapy.
Genetically engineered glucocerebrosidase (recombinant imiglucerase) is infused intravenously. Since the modified enzyme substrate has a relatively long half-life, two-week infusions are sufficient. The substrate is taken up by the macrophages and can thus catalyze the breakdown of glucocerebroside. Enzyme replacement therapy is the standard therapy for the non-neuronopathic and chronic neuronopathic forms of Gaucher's disease and brings about a gradual improvement in the symptoms characteristic of Gaucher's disease.
In addition, in the case of a mild course of Gaucher's disease, a therapeutic approach is pursued in which the glucocerebroside accumulation is partially inhibited by the orally administered active ingredient miglustat (substrate reduction therapy). Due to severe side effects, this drug is only used in people with Gaucher's disease for whom enzyme replacement therapy is not indicated.
In addition, accompanying measures can be taken to reduce or eliminate the respective symptoms. For example, if the bones are severely impaired, additional orthopedic measures, including joint replacement, may be necessary.
Outlook & forecast
Without treatment, all Gaucher disease types show a slowly progressive course. During treatment, the prognosis depends on the type of disease present. Gaucher's disease type I is usually well treatable with early and timely therapy. The quality of life here is primarily impaired by the bone and joint changes. Growth disorders and bone crises are often the focus of childhood. Some people need a wheelchair after fractures and necrosis of the femoral head.
In the case of Gaucher's disease type II (acute neuronopathic form), however, the prognosis is poor due to the pronounced involvement of the nervous system. Despite therapy, most affected children die within the first two years of life. Gaucher's disease type III (chronic neuronopathic form) is treatable, but can be associated with noticeable mental impairments and a reduced life expectancy. However, there are not enough data from clinical studies for this type so that a final assessment is not possible.
All Gaucher disease sufferers have an increased risk of bleeding complications and ruptured spleen during the entire course of the disease. A final cure can only be achieved through gene therapy. Fetal gene therapy for type II has been successfully carried out by British researchers, at least on mice. It is not yet possible to say whether and when such a therapy will be available for Gaucher disease sufferers.
prevention
There Gaucher's disease is a genetic lipid storage disease, it cannot be directly prevented. However, a heterozygous test and prenatal diagnosis can be used during pregnancy to determine whether the child will be affected by Gaucher's disease.
Aftercare
As a genetic disease, Gaucher's disease is still incurable today. Those affected are dependent on taking medication for life. Once patients have been diagnosed and are ready for therapy, regular follow-up is required to monitor the success of the treatment. As a rule, the doctor calls on those affected to take a blood sample once a quarter in order to check the most important values.
Depending on the severity of the disease and its course, thorough examinations in a specialized Gaucher competence center are also required every six to twelve months. Depending on the type and stage of the disease, the disease may lead to severe pain and movement disorders and symptoms of paralysis. Experts advise keeping a pain diary and starting pain therapy in order to largely preserve the quality of life.
In addition, exercises that can be easily integrated into everyday life help to improve and maintain mobility. In principle, a healthy and balanced diet is also highly recommended for Gauchers syndrome. There is an increased need for calcium and iron.
That is why dairy products and oatmeal, lentils or broccoli are a valuable part of the menu. If you feel good, regular exercise is also useful. However, those affected should avoid contact sports, as this disease has an increased risk of a ruptured spleen.
You can do that yourself
Gaucher's disease patients can do a lot themselves to improve their quality of life. Active coping with illness and self-responsible handling of the illness strengthen self-confidence. Those affected do not get into a depressive downward spiral that quickly. It is important to first accept the disease. Rebellion, on the other hand, only costs unnecessary strength.
It is also advisable to research as much as possible about the clinical picture. The more knowledge there is, the less fear and insecurity become. Open questions can be discussed with the trusted doctor. It is also advisable to visit a self-help group. In exchange with other patients, those affected feel understood and no longer so lonely. It is also advisable to seek psychotherapeutic help.
A healthy, balanced diet is generally recommended. Gaucher's disease patients can benefit from this in a special way. Calcium strengthens muscles and bones. Iron helps against anemia. Natural foods such as dairy and whole grain products, fish, spinach, nuts and legumes contain many times the nutrients needed. In addition, dietary supplements can be taken to meet daily needs. Of course only after consultation with a doctor.
Exercise can also have positive effects on health. Most sports are easy to do. To be on the safe side, the attending physician should be consulted.
.jpg)











.jpg)