The Sialic Acid Storage Disease is a very rarely occurring, genetic lysosomal storage disease in which the protein sialin is incorrectly encoded. Sialin is a transmembrane protein that normally removes monosaccharides such as sialic acid, which arise from the enzymatic breakdown of glycoproteins and other substances, from the lysosome. The reduced functionality of the sialin leads to an accumulation of sialic acid in the lysosomes, i.e. to sialic acid storage disease.
What is sialic acid storage disease?

© LIGHTFIELD STUDIOS - stock.adobe.com
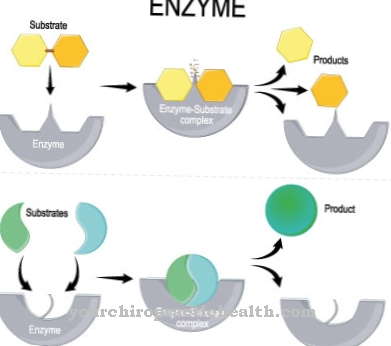
The term sialic acid includes derivatives of neuraminic acid that are N- or O-acetylated. Sialic acids are building blocks of glycoproteins, glycosaminoglycans and other substances and are released through their enzymatic breakdown within special lysosomes.
Normally, the sialic acids are channeled out of the lysosome through the membrane of the lysosome with the aid of the anion transport protein sialin. Characteristic of the Sialic Acid Storage Disease, which are also called Neuraminic acid storage disease is a functional restriction or a total loss of function of the transport protein sialin.
The sialic acids produced in the lysosomes can be channeled out of the lysosomes, so that there is an excessive accumulation of the sialic acids in the lysosomes. An infantile and an adult form of lysosomal sialic acid storage disease are known. The adult form is also known as Salla's disease.
causes
The causes for the loss of function of the transmembrane protein sialin go back to a defect or a mutation of the SLC17A5 gene in the infantile as well as in the adult form. The gene is on chromosome 6, on the gene locus q14-q15. All known mutations of the gene lead to an incorrect coding of the protein sialin.
Although it is accepted as a building block by the metabolism, it does not have the ability to discharge sialic acid through the membrane of the lysosome. The wrong coding of the sialin gives the protein a different tertiary structure, which is associated with the loss of functionality as a transport protein. Different gene mutations lead to the infantile or adult form of the sialic acid storage disease.
Symptoms, ailments & signs
Infantile sialic acid storage disease is the one with the less favorable prognosis of both forms. The first signs and symptoms manifest themselves before birth. In the prenatal diagnosis by ultrasound, a so-called hydrops fetalis can usually be found, an accumulation of tissue fluid that extends over large parts of the fetus.
Postnatally, enlargement of the liver and spleen is symptomatic and facial features are mostly rough. Life expectancy in the infantile form of neuraminic acid storage disease is usually only a few years. Salla disease or the adult form of sialic acid storage disease is already noticeable in infancy.
This happens through muscular hypotension and a horizontal nystagmus, in which the eyes make jerky horizontal movements. Those affected show mental disabilities and mostly do not have a language ability. The prognosis for life expectancy is slightly more favorable than that of the infantile form of the disease. Those affected usually reach adulthood.
Diagnosis & course of disease
In people affected by sialic acid storage disease, an increased concentration of sialic acid in the urine and an increased excretion rate of polysaccharides are symptomatic. Free sialic acid can be detected in fibroblasts and amniotic cells. A genetic test, which can also be carried out prenatally, serves as the final certainty.
In severe cases of sialic acid storage disease, bone malformations, ascites (ascites), severe movement disorders, spastic cramps and severe mental disabilities become noticeable soon after birth. The severe forms of the disease lead to death in early childhood.
With moderate courses - as they are typical for Salla's disease - those affected usually reach adulthood. Severe intellectual disabilities, spastic convulsions and severe psychomotor restrictions are symptomatic of these forms of disease. Although the disease is very rare worldwide, it has a regional focus in northern Finland, where every 40th heterozygous carrier is one of the corresponding gene mutations.
Complications
In most cases, sialic acid storage disease can be recognized and diagnosed before birth. Those affected usually suffer from a significantly enlarged spleen and liver. This can lead to pain in the respective regions. The facial features of the patients are often coarse, so that teasing or bullying can occur, especially at a young age.
Many children suffer from psychological complaints or depression. Furthermore, those affected often suffer from intellectual disabilities and thus also from restrictions in everyday life. They are therefore often dependent on the help of other people in their everyday life and cannot easily do many everyday things. Sialic acid storage disease can also lead to language problems, so that communication between those affected is considerably restricted.
Due to the sialic acid storage disease, the life expectancy of the patient is usually also considerably reduced. Most of those affected only reach adulthood with it. Since causal therapy is not possible, only the symptoms can be limited. However, there are no particular complications. The sialic acid storage disease cannot be prevented.
When should you go to the doctor?
Sialic acid storage disease usually always requires medical examination and treatment. Since this is a hereditary disease, a genetic test should also be carried out if you want to have children in order to prevent the disease from being passed on to your descendants. Since this does not lead to self-healing, the person affected with sialic acid storage disease is in any case dependent on lifelong treatment by a doctor.
A doctor should be consulted if the patient has a significantly enlarged spleen or liver. In most cases, these symptoms are discovered by chance through an ultrasound examination, although pain in the sides of the body can also indicate these symptoms. In some cases, problems with speaking or a number of intellectual disabilities point to sialic acid storage disease.
The first examination of this complaint can be carried out by a general practitioner. Further treatment is usually carried out by various specialists and is always based on the exact symptoms.
Treatment & Therapy
Since the sialic acid storage disease is based on a genetic defect, a causal therapy is not possible. There is no way to replace the functionally impaired transmembrane and anion transport protein with functional sialin. The possible therapies are (still) limited to treating the symptoms and ensuring that the patient experiences as little pain as possible.
In the case of some other lysosomal storage diseases such as Hunter's disease and Gaucher's disease, the infusion of genetically engineered enzymes has already shown positive results. The procedure is known as enzyme replacement therapy and is effective on internal organs and connective tissue. Because of their size, the artificially supplied enzymes cannot cross the blood-brain barrier, so that such therapies have no influence on developments in the CNS.
One form of therapy that is currently still in its infancy is so-called gene therapy, which is intended to lead to a targeted exchange of the mutated gene sequence. In principle, this is a technique that is used in a positive sense in the body's own gene repair or can also have negative effects in the form of certain pathogenic germs.
You can find your medication here
➔ Medicines for painprevention
There are no preventive measures that could prevent the outbreak of sialic acid storage disease, because it is a genetically determined, systemically effective disease that is already created prenatally.
In families in which, for example, cases of Salla's disease have become known, if they wish to have children, a genetic examination can be carried out to determine whether the potential parent is a carrier of one of the known gene mutations without being sick themselves. In the positive case, a consultation should take place in which the risks that exist for the child in realizing the desire for children should be explained.
Aftercare
As a rule, patients with sialic acid storage disease do not have any special options for direct follow-up care. For this reason, this disease should be diagnosed and treated early by a doctor so that there are no complications or other symptoms in the further course.
The earlier a doctor is consulted, the better the further course of this disease is usually, so that a doctor should be contacted as soon as the first signs and symptoms appear. Sialic acid storage disease usually cannot heal itself. Most patients with sialic acid storage disease are dependent on taking various medications in order to relieve the symptoms of the disease permanently and properly.
Regular consumption and the prescribed dosage must be observed in order to counteract the symptoms correctly. Regular check-ups by a doctor are also very important in order to identify and treat other damage at an early stage. Because of the illness, some of those affected are also dependent on the help and support of their own families. This disease may also reduce the patient's life expectancy.
You can do that yourself
Since the disease is based on a genetic defect, the person affected cannot take any options that contribute to a cure for the health disorder.The focus of the self-help measures is to be directed towards improving daily processes and wellbeing.
It must be taken into account that a large number of patients suffer from impaired cognitive performance. Therefore, they are often dependent on the help and support of relatives or medically trained nursing staff. When coping with the disease, parents or other family members in particular must make sure that they take their own limits into account and do not give themselves up on care and support. The development of elements to improve the joy of life is immensely important for everyone involved. A stable social environment also helps with adversity.
Since the disease has a negative effect on the development of some organs, this should be taken into account in everyday life. A healthy diet, optimal sleep hygiene and regular daily routines help to cope with the disease. They bring security and stability. Since the communication of the patient is limited, possibilities should be developed so that a good exchange of all those involved can take place in everyday life. Mutual understanding, calm and the avoidance of hectic pace are beneficial for further development.

.jpg)
.jpg)
.jpg)
.jpg)









.jpg)