Sphingolipids belong to the building blocks of the cell membrane alongside glycerophospholipids and cholesterol. Chemically, they are derived from sphingosine, an unsaturated amino alcohol with 18 carbon atoms. Mainly the nervous system and brain are rich in sphingolipids.
What are sphingolipids?
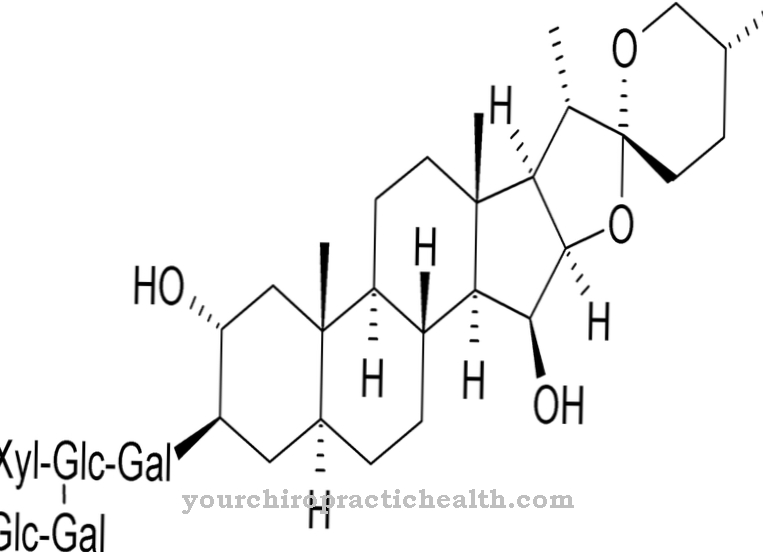
All cell membranes contain glycerophospholipids, cholesterol and sphingolipids. Sphingolipids consist of the basic structure sphingosine, on whose amino group a fatty acid is esterified. Sphingosine is an amino alcohol that contains a chain of 18 carbon atoms. The sphingolipids can be divided into three groups.
These are the ceramides, the sphingomyelins and the glycosphingolipids. Ceramides are the simplest sphingolipids. Here, sphingosine is esterified with a fatty acid. An amphiphilic double structure with double lipid layers is formed. Amphiphilia is created by the two hydrocarbon tails, each pointing in the opposite direction. Sphingomyelins are esterified on the hydroxyl group of the sphingosine skeleton with a phosphoric acid, which in turn is esterified either with an alcohol or with choline.
Finally, glycosphingolipids have a glycosidic bond with a sugar residue on the hydroxyl group of the sphingosine skeleton. The cerebrosides are monohexoses, while the gangliosides contain an oligosaccharose that is glycosidically bound.
Function, effect & tasks
The sphingolipids fulfill different functions. Their structure plays a major role here. The simplest sphingolipids, the ceramides, are particularly involved in the structure of the horny layer of the skin. Due to their amphiphilicity, they can form a lipid bilayer that protects the skin from drying out.
In addition to this function, however, ceramides also fulfill numerous other tasks. These include signal transmission or tasks in controlling cell division. In addition to glycerophospholipids and cholesterol, sphingomyelins are responsible for the fluidity of cell membranes and the transport of substances. The functions of glycosphingolipids are also diverse. As already mentioned, the glycosphingolipids contain a glycosidically bound hexose or an oligosaccharide on the hydroxyl group of the sphingosine skeleton. As a result, they have a hydrophobic ceramide component and a hydrophilic sugar component.
This sugar component is placed on the surface of the cell membrane, which can result in cell-cell interactions through cell adhesion. Therefore they are of great importance for the signal transmission of nerve cells. Glycosphingolipids are also largely responsible for the cell interactions of the other cells.
Education, occurrence, properties & optimal values
The biochemical synthesis of the sphingolipids takes place in the endoplasmic reticulum and in the Golgi apparatus. From there they are transported to the membranes with the help of vesicles. The sphingolipids are converted even further in the membranes so that they can fulfill their numerous tasks there. All cell membranes contain sphingolipids. However, their concentration is particularly high in the brain cells and nerve cells. This is especially true for the gangliosides. For example, they make up six percent of the lipids in the gray matter of the brain.
The cerebrosides esterified with a hexose are found to a greater extent in the brain and liver. In the brain, the glycosidically bound sugar is mostly galactose, while the cerebrosides in the liver mainly contain glucose. The simplest sphingolipids, the ceramides, are particularly found in the skin and there in the stratum corneum (horny layer). As already mentioned, due to their amphiphilia, they can form a barrier there, which protects the skin from water loss. Of course, the ceramides are also found in the other cell membranes, as they have to fulfill even more functions in terms of signal transmission and cell control. Sphingomyelin is also found in all cell membranes. However, its highest density is again in the neurons.
Diseases & Disorders
So-called storage diseases can occur in connection with sphingolipids. These are mostly genetic diseases that are characterized by a missing or inactive enzyme. As a result, the breakdown of the corresponding sphingolipids is no longer possible. The sphingolipid accumulates in the cell and leads to its destruction.
Many storage diseases are fatal after years of suffering. Typical lipid storage diseases include Tay-Sachs syndrome and Niemann-Pick disease. In the course of these diseases, sphingolipids are accumulated in the cells. Tay-Sachs disease is caused by an autosomal recessive hereditary mutation in a gene that codes for the enzyme β-hexosaminidase A. This enzyme is responsible for breaking down the ganglioside GM2. As a result of its failure, ganglioside GM2 accumulates especially in the nerve cells. The patient suffers from central nervous and motor disorders as well as mental retardation. This disease leads to death within the first three years of life.
Niemann-Pick disease is also inherited as an autosomal recessive trait. In this disease, sphingomyelins accumulate in the cell membranes. Endothelial, mesenchymal and parenchymal cells are particularly affected. The esterification of cholesterol is also disturbed, so that this too is deposited in the cells. There are different forms of disease in Niemann-Pick disease. These depend on the respective activity of the sphingomyelinase. In the classic form of the disease, symptoms begin during the first year of life. Death usually occurs before the end of the third year of life. If the disease starts later, symptoms will develop more slowly. Then Niemann-Pick disease is characterized by increasing liver and spleen enlargement, cramps, movement disorders, muscle tremors and mental retardation.
Another sphingolipidosis is Gaucher's disease. Gaucher's disease is Gaucher's disease. Glucocerebrosides are constantly deposited in various body cells such as the liver, spleen, bone marrow and macrophages. This disease usually leads to death at the age of two. Gaucher's disease, however, can be treated by replacing the enzyme glucocerebrosidase.