The I cell disease is a lyosomal mucolipidosis. The cause of the storage disease is a mutation of the GNPTA gene with the q23.3 gene locus on chromosome 12. Symptomatic treatment is mainly carried out by administering bisphosphonates.
What is I cell disease?

© red150770 - stock.adobe.com
Storage diseases are characterized by the deposition of various substances in cells and organs of the human body. It is a heterogeneous group of diseases that can be divided into several sub-forms. In addition to glycogenoses, mucopolysaccharidoses and lipidoses, medicine differentiates, depending on the substance deposited, sphingolipidoses, hemosideroses and amyloidoses.
Lysosomal storage diseases affect the lysosomes. These are tiny, membrane-coated cell organelles in the eukaryotes. Lysosomes are formed by the Golgi apparatus and are equipped with hydrolytic enzymes and phosphatases. With the help of their enzymes, they should primarily digest foreign substances and the body's own substances.
I-cell disease is a lyosomal mucolipidosis with two different subtypes. Leroy and DeMars first documented the disease in the 1960s, pointing to its similarity to mucopolysaccharidosis type I, known as Hurler's disease. The name of the disease comes from the fibroblast inclusions, the so-called inclusion cells, in the patient's skin.
causes
The cause of the I-cell disease lies in a lack of activity of the N-acetylglucosaminyl-1-phosphotransferase. The restricted activity of this enzyme prevents a large part of the lysosomal enzymes from entering the interior of the lysosome. The regulation of the lysosomal enzymes is shaped by the activity of the phosphotransferase.
It enables the synthesis of a sorting signal in a healthy organism. This process is disturbed in I-cell disease. Therefore, there is no labeling with mannose-6-phosphate. For this reason, the lysosomal enzymes are no longer properly sorted and migrate into the extracellular matrix in an uncontrolled manner via the plasma membrane.
The reason for this is a mutation in the GNPTAB gene. It removes the functionality of the N-acetylglucosaminyl-1-phosphotransferase and thus the ability to catalyze the synthesis of mannose-6-phosphate. The transport of lysomal enzymes is so disturbed. The N-acetyl-glucosamine-1-phosphotransferase consists of the subunits alpha, beta and gamma. They are encoded on two genes.
The hereditary I-cell disease affects the GNPTA gene on chromosome 12. There is a mutation in the q23.3 gene locus. For the rare disease, an incidence of around 0.3: 100,000 is given. Inheritance is subject to autosomal recessive inheritance. Both parents must therefore carry the defective gene in order to pass the disease on.
Symptoms, ailments & signs
In most cases, the symptoms of I-cell disease can be observed immediately after birth or a few months later at the latest, and their characteristics are similar to those of Hurler's syndrome. Unlike patients with Hurler syndrome, those with I-cell disease do not show any mucopolysaccharide excretion.
The individual symptoms of the disease are subject to a large number of variations. Kornfeld and Sly summarize clinical features of the skeleton, internal organs, eyes, skin, central nervous system and face. The skeleton is so often affected by kyphoscoliosis and hip dislocations.
Club feet, joint contractures and deformities of the vertebrae can also be present. The same applies to short stature and dysostosis multiplex. The disease can manifest itself in the internal organs in the form of hepatosplenomegaly and cardiomegaly or heart disease. The patient's face has coarsened features.
Exophthalmos, hyperplastic gums or scaphocephaly are typical symptoms. Also characteristic are an open mouth and a deeply sunken nose. The eyes of those affected often have corneal opacities or swollen eyelids. The skin is thick and coarse, with severe psychomotor or mental retardation in the central nervous system.
Diagnosis & course of disease
The first suspected diagnosis of I-cell disease can be made by visual diagnosis based on the anamnesis. A biochemical determination of the lysosomal enzyme activity in the serum can be used to confirm the diagnosis. This determination reveals an absurd relationship between intra- and extracellular activity.
The activity of phosphotransferase in the fibroblasts can also be determined to confirm the diagnosis. The inclusions correspond to either mucopolysaccharides, lipids or oligosaccharides. Molecular genetic diagnostics can dispel any remaining doubts. If there is an appropriate history, the disease can also be diagnosed as part of prenatal diagnosis.
Because of the low prevalence, prenatal work-up is actually only recommended if there is a family disposition. The course of the disease depends on the symptoms in the individual case and is not directly predictable. Most patients, however, barely survive the age of ten. However, milder forms of development are not entirely excluded in individual cases.
Complications
I-cell disease can lead to various complications and complaints. These are recognized late, however, so that the I-cell disease can only be diagnosed late. The symptoms are relatively inconsistent, which often makes treatment difficult. This usually leads to discomfort and malformations of the skin, eyes and internal organs.
In the worst case, the affected person can go blind or die directly from organ failure. Furthermore, there is a pronounced short stature and also heart problems. The eyelids are often swollen and there is decreased intelligence and mental retardation. It is not uncommon for the person affected to be dependent on the help of other people in everyday life due to the retardation in order to cope with it.
The patient's quality of life is greatly reduced by the I-cell disease. As a rule, there are no particular complications in treating the disease. Medicines and psychological treatments are used that can alleviate the symptoms. However, a complete and causal treatment of this disease is not possible. Life expectancy is reduced by the disease.
When should you go to the doctor?
I-cell disease is usually diagnosed immediately after the child is born. Whether further treatment measures are necessary depends on the type and severity of the symptoms. Minor malformations do not necessarily have to be treated. Club feet and deformities of the vertebrae, on the other hand, are serious malformations that must be treated surgically and with medication. Parents should consult a specialist immediately if the doctor responsible at the maternity hospital has not already done so.
If an accident or fall occurs as a result of the complaints, the child must be taken to hospital or the parents should call the emergency services immediately. In the case of severe malformations, which may also affect the child's psyche later in life, a therapist should be consulted to accompany the medical treatment. The I-cell disease therefore always requires a medical examination. The right contact person is the pediatrician or a specialist in hereditary diseases. In the event of visual disturbances, an ophthalmologist should be consulted.
Doctors & therapists in your area
Treatment & Therapy
The I-cell disease is considered incurable. A causal therapy therefore does not exist. Treatment is only symptomatic and supportive. Psychotherapeutic care for affected families makes up a large part of supportive therapy. Symptomatic therapy depends on the individual case. The bone symptoms are often treated by giving bisphosphonates.
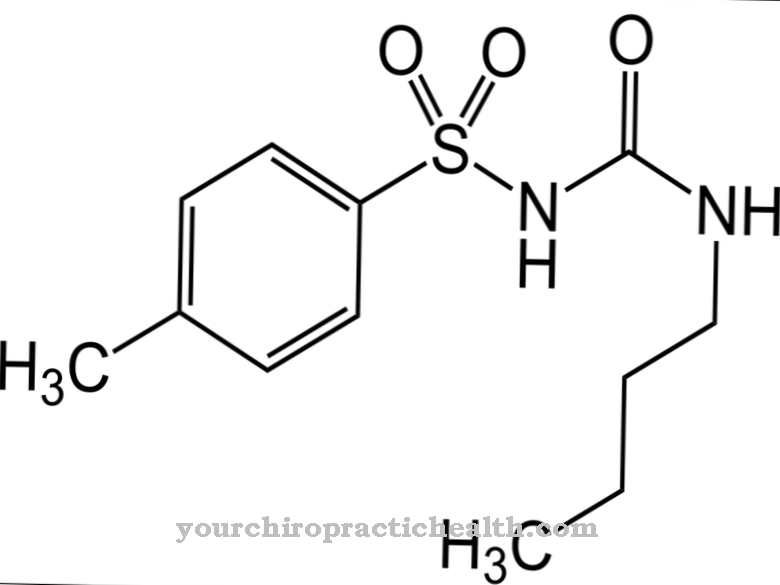
These drugs are known from the treatment of osteoporosis and have a high affinity for the bone surface. Especially in the region of the resorption lacunae, they attach to the bones. In doing so, they inhibit the bone-degrading osteoclasts and in this way reduce bone resorption. The drugs are pyrophosphate analogs with a carbon-containing P-O-P bond.
An enzymatic hydrolysis does not take place on them. The aminobisphosphonates are among the most recent of these substances. In addition, alendronate, clodronate, etidronate, ibandronate, pamidronate and risendronate are approved in Germany from the same drug group. The same goes for tiludronate and zoledronate.
In addition to these drugs, bone marrow transplants can also be used to treat I-cell disease. The success of this treatment has only been limited in previous cases. Gene therapies are now being investigated as a new therapeutic approach for gene defects. Gene therapies have shown initial success in animal models. So far, they have not been able to be used in practice on humans. However, this relationship will presumably change in the future.
You can find your medication here
➔ Medicines for painOutlook & forecast
I-cell disease is an inherited disease that has not yet been treated symptomatically. The prognosis is accordingly negative. Although the symptoms can be significantly reduced by early therapy, the I-cell disease almost always takes a serious course.
The short stature and the damage to the internal organs and the head already reduce life expectancy considerably. Furthermore, malformations in the face, skin and eyes can reduce life expectancy, but primarily also the quality of life of the person affected. Some of those affected reach the age of 40 or 50, but most of them die in childhood or adolescence.
If the I-cell disease is not treated, the sick often die in the first few years of life. The prognosis is therefore rather negative. Nevertheless, the prospect of a relatively symptom-free life is given if the patient is treated as part of a comprehensive therapy and, if necessary, is placed in an institution for physically disabled people. Physiotherapy and therapeutic measures can significantly improve the patient's wellbeing in the long term.
prevention
I-cell disease can only be prevented by a molecular genetic test before family planning. As part of the prenatal diagnosis, expectant parents can also decide to terminate the pregnancy.
Aftercare
In most cases, those affected with I-cell disease have no or very few follow-up measures available. The disease must be recognized by a doctor as early as possible so that a further worsening of the symptoms can be prevented. Since this is a genetically determined disease, a genetic examination and consultation should always be carried out first in the case of a desire to have children in order to avoid the inheritance of the I-cell disease to descendants.
Most patients are dependent on various medications for this disease. It is important to ensure that the dosage is correct and that the medication is taken regularly. If anything is unclear, there are side effects or if you have any questions, you should always consult a doctor first.
Likewise, many sufferers need psychological support with this disease, although loving discussions with parents or relatives can have a positive effect on the course of the disease. A person affected needs the help and support in everyday life from their own family. In many cases, the I-cell disease significantly limits or reduces the life expectancy of the person affected.
You can do that yourself
Patients suffering from I-cell disease can resort to various conservative and alternative treatment methods. Conservative therapy focuses on relieving symptoms and ailments.
The use of aids such as crutches or orthopedic insoles can slow down the course of the respective malformations and thus also reduce the pain. Medication helps relieve pain and can be supplemented with alternative measures such as massage or acupuncture. Alternative therapies should be discussed with the responsible doctor beforehand. The doctor may be able to refer the patient directly to a homeopath or give further tips on how to treat the respective symptom.
Since the I-cell disease is usually fatal despite all treatment options, therapeutic advice should be sought. Not only those affected should work through their fears. The relatives and friends usually also need support in dealing with the disease and its possible negative outcome. Participation in a self-help group is also an option for the patient and their relatives. Contact with other sufferers helps to accept the disease, and often other sufferers can also suggest further treatment measures and strategies for daily living with I-cell disease.
.jpg)

.jpg)



.jpg)
.jpg)
.jpg)



.jpg)