There are 45 different in total lysosomal storage diseases, which are a heterogeneous group of congenital metabolic diseases. People who suffer from any of these diseases have a genetic defect. All storage diseases have one thing in common: a certain enzyme is either absent or only partially functional.
What is lysosomal storage disease?

© designua - stock.adobe.com
These congenital storage diseases are rare, as fewer than five in 10,000 people are affected. The various diseases have a very different course, and the symptoms can vary widely.
The most famous forms of lysosomal storage disease are Fabry's disease, Gaucher's disease, Pompe disease and mucopolysaccharidosis (MPS). They are often referred to as "orphans of medicine" because the path to a specific diagnosis and suitable therapy can be very long. Sometimes it can take years for those affected to find out what is happening to them.
causes
Lysosomal storage diseases are characterized by certain forms of hereditary metabolic diseases. The patient lacks an important enzyme that ensures that the metabolic balance runs smoothly. In the less pronounced form, this enzyme is at least not present in sufficient quantities.
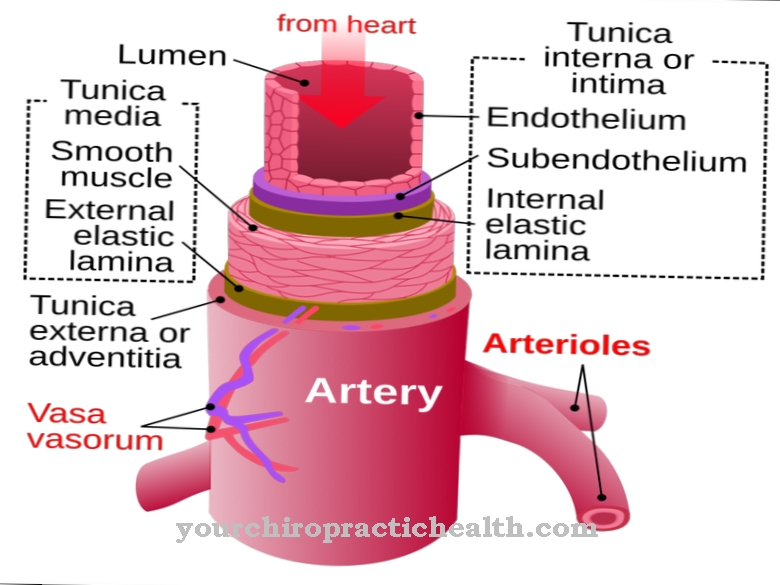
The task of enzymes is to dispose of pollutants and waste substances that accumulate in the human organism via the metabolism via the lysosomes, or to process them again in such a way that symptoms do not occur.
If there is an enzyme deficiency, this smoothly functioning disposal cycle is no longer guaranteed. The harmful substances settle in the cells and disrupt the metabolic cycle. In the initial phase, the disturbances do not have a noticeable effect, there are only a few restrictions. However, if this metabolic disorder remains untreated as a result of an enzyme deficiency, the symptoms multiply because the cells become very enlarged.
Symptoms, ailments & signs
In the worst case, these go under. The consequences are damage to the bones, the nervous system, spleen, kidneys, muscles or heart. Due to the reduced or absent enzyme activity, Fabry disease causes fat (globotriaosylceramide, Gb3) to be stored in the cells. These unwanted deposits can lead to severe pain in the toes or fingers, stroke, and kidney damage.
Diagnosis & course of disease
This clinical picture affects different systems at the same time: blood vessels, kidneys, heart and nervous system. The autosomal recessive Gaucher disease causes a mutation of the enzyme "beta-glucocerebrosidase" and leads to an accumulation of substrate within the cells, especially in the macrophages (phagocytes) that belong to the reticulo-endothelial system. The blood count changes, the liver and spleen are enlarged, and the bones hurt.
The disease is progressive and is mostly ethnic, since it occurs in most cases in people of Jewish descent. Pompe disease is also known as "acid maltase deficiency". The clinical picture belongs to the group of glycogenesis type II. The affected persons lack the enzyme "alpha-1,4-glucosidase" (acid maltase) or it is not available in sufficient quantities. Due to the impaired glycogen breakdown in the muscles, patients suffer from the destruction of the muscle cells in the form of sugar storage.
Mucopolysaccharidosis type I (MPS), also known as Hunter's disease, has various clinical causes. Hurler's disease is the most severe form and Scheie's disease is at the end of clinical pathogenesis. There are transitions of different characteristics between these two forms of progression. The most striking property is the impaired breakdown of carbohydrates that accumulate in the lysosomes of the cells.
Hunter disease patients may experience short stature, enlarged spleen and liver, gross features, thickened skin, enlarged tongue, and difficulty breathing. In addition, the skeleton is often changed in the area of the pelvis, spine, hand bones and skull. Umbilical and [[inguinal hernias] are possible.
Complications
In most cases, symptoms or complications appear very late in this disease. Because of this, it is diagnosed late, making early treatment in most cases impossible. Without treatment, various complaints and damage to the internal organs occur as the disease progresses.
The kidneys, liver and spleen are particularly affected. The heart can also be affected by this disease, which can lead to cardiac death in the worst case. Furthermore, damage to the kidneys occurs and those affected often suffer from pain in the toes or fingers. Paralysis can also occur if the brain has been damaged by this disease. The liver and spleen can be enlarged and cause severe pain as well.
It is not uncommon for the affected person's bones to be brittle and also painful. Treatment of this disease is proving difficult. In many cases, the life expectancy of the person affected is significantly reduced. There are usually no particular complications when using medication. However, a positive course of the disease cannot be guaranteed in every case.
You can find your medication here
➔ Medicines for painWhen should you go to the doctor?
Hair loss, joint problems and organ disorders are possible signs of lysosomal storage disease. A visit to the doctor is recommended if the symptoms keep recurring or appear suddenly without a cause being found. If the symptoms are related to a previously diagnosed enzyme defect or other serious illness, the responsible doctor should be consulted. An untreated storage disease can lead to dementia, infertility, neuropathies and other complications, some of which are life-threatening. Therefore, all conceivable symptoms should be examined, even if there is no specific suspicion.
The symptoms of lysosomal storage disease can appear in phases or develop insidiously, but always require examination and treatment. Affected people are best to speak directly to their family doctor or an internist. The actual therapy usually takes place in a specialist clinic for internal diseases, although physiotherapy or psychotherapy can be connected depending on the symptoms. In particular, therapeutic measures are indicated due to the often negative course of the disease.
Therapy & Treatment
Depending on how early an adequate diagnosis is made, these hereditary diseases can be treated very well with enzyme replacement therapy, so that the people affected have far fewer complaints and thus a better quality of life. This replacement therapy is used according to the clinical picture.
People who suffer from Gaucher's disease lack the “enzyme ß-glucocerebrosidase”, which is produced biotechnologically and infused into the patient's body. Lysosomes act efficiently and are able to absorb substances from their immediate environment. For this reason, the artificially used enzymes are modified in such a way that they can be supplied to the lysosomes in an ideal way.
The macrophages (phagocytes) break down the glucocerebrosides that have accumulated in the cells. This therapy can be compared with insulin therapy for diabetes mellitus, with the difference that it is not a missing hormone but an enzyme that is not supplied. The body regularly breaks down all substances, including the supplied artificial enzyme.
Because of this regular breakdown of the substance, patients have to undergo this infusion therapy regularly until the end of their life. The enzyme replacement therapy does not act symptomatically, but directly combats the cause of the hereditary disease. Doctors call this therapy causal. The principles of therapy are to be used for all four of the aforementioned common storage diseases.
Pompe patients are also treated with infusion therapy. In this disease, the non-existent enzyme "acid alfa glucosidase" is supplied and helps break down glycogen that has accumulated in the lysosomes of the muscles. In patients with the disease type “mucopolysaccharidosis type I” the lysosomal enzyme “alpha-iduronidase” is not present or is not present in sufficient quantities. It is one of the rarest storage diseases in which sugar molecules accumulate in organs and tissues.
If the process is normal, the enzyme breaks down mucopolysaccharides. The sugar molecules are long-chained and are involved in the development of supporting and connective tissue, for example bones, skin, joint fluids and cartilage. If the normal course of degradation is disturbed due to the lack of enzyme, pathological glycosaminoglycans (GAG) accumulate in the individual cells. Future therapy options are aimed at taking tablets.
Outlook & forecast
The prognosis for storage disease is poor. A genetic disposition was found to be the cause of the health disorder. Legal requirements prohibit physicians and scientists from changing human genetics. For this reason, the disease remains lifelong and has no prospect of recovery.
The attending physician concentrates on treating the symptoms that arise. If left untreated, various complaints will increase over time. The bone system is damaged and problems of the organs arise. In the worst case, the internal organs will malfunction and ultimately their function will fail. This threatens the person concerned with premature death.
The challenge of the disease lies in the diagnosis. In a large number of patients, noteworthy and strongly perceptible complaints only occur later in life. As a result, the genetic disorder remains unnoticed for a long time and early treatment of the disease is difficult. The later a diagnosis is made, the more unfavorable the further course is. At an advanced stage of the disease, the internal organs or joints are already severely damaged. Surgical interventions are required and if the disease progresses unfavorably, only one donor organ can save the life of the person affected. Early treatment is therefore essential for an improved prognosis.
prevention
Since it is a congenital genetic defect that prevents the expression of an enzyme, this disease cannot be treated preventively. However, the latest genetic engineering achievements could provide an approach in this field.
Aftercare
With this disease, people suffer from a number of different complications and ailments. As a rule, these all have a very negative effect on the quality of life of the person affected, so that a diagnosis should be made very early on.The earlier a doctor is consulted, the better the further course of this disease is usually.
The severity of this disease can be very different, so that a general prediction is often not possible. Those affected suffer from severe damage to the internal organs. This primarily affects the kidneys and heart, so that the child can die in the first few days if the symptoms are not corrected in time. There are also deposits of fat in different parts of the body.
The fingers and toes are particularly affected, which can lead to significantly reduced aesthetics for the person affected. As a rule, damage to the kidneys and the brain occurs in the further course, so that the person affected dies as a result of this damage. The parents and relatives also often suffer from depression or other mental disorders due to the illness.
You can do that yourself
Lysosomal storage diseases very often require intensive medical care. Often there are not enough opportunities for self-help. The parents of affected children often experience severe stress in their home environment because their child needs constant care and attention.
The clinical pictures of the individual storage diseases are different. There are both easy and very difficult forms. One example is Gaucher's disease. Parents' help is often limited to feeding the severely disabled child. In milder cases, life expectancy can be almost normal. Nonetheless, constant medical supervision is necessary to avert possible complications. Regular physical activity is one of the accompanying therapies that can also be carried out at home. Furthermore, a careful cancer screening examination must be arranged. This requires constant visits to the doctor with their child from the parents. The same applies to other lysosomal storage diseases.
In the case of some illnesses, in addition to physical disabilities, mental limitations can also occur, which require special support. In milder forms of certain diseases, such as Hunter's disease, initially only skeletal changes and facial dysmorphisms occur. Here, however, the affected patient is often able to lead an independent life. However, constant medical examinations are also required here in order to rule out possible complications such as heart failure or respiratory diseases. The patient can deal with psychological stress caused by physical deformations through psychological counseling.
.jpg)
.jpg)





.jpg)
.jpg)
.jpg)



.jpg)