Of the Pompe disease is a glycogen storage disease of unpredictable course. The symptoms are characterized by progressive muscle weakness. In therapy, successes through the artificial administration of the causative enzyme have meanwhile been observed.
What is Pompe disease?

© designua - stock.adobe.com
Storage diseases are a heterogeneous group of diseases in which various substances are deposited in organs or cells of the organism. The tissues or organs usually lose their function due to the deposits. The substances deposited can be of various types. Depending on the substance, a distinction is made between glycogenoses, mucopolysaccharidoses, lipidoses, sphingolipidoses, hemosideroses and amyloidoses. In glycogenosis, glycogen is stored around body tissue.
The stored carbohydrates can no longer be broken down at all or only incompletely or can be transformed into glucose. The cause is usually an enzyme deficiency in the enzymes that are involved in converting glucose in the body. A glycogen storage disease is that Pompe diseasewho too Pompian disease or Acid maltase deficiency is called. The disease affects the lysosomal α-glucosidase or acid maltase on the GAA gene.
In the healthy body, the enzyme breaks down the long-chain polysaccharides of the lysosomes into glucose. In humans, the enzyme is present in all tissues. The metabolic disease is still mainly noticeable in the muscles and is therefore also included in myopathies. The rare disease is named after the Dutch pathologist Pompe, who first described the phenomenon in 1932. In 1954 G.T. Cori's disease as a glycogen storage disease type II.
causes
In the 1960s, H.G. Hers the lack of lysosomal α-glucosidase as a causal link in Pompe disease. This deficiency arises on the basis of a primarily causal genetic defect that affects the enzyme α-1,4-glucosidase. This enzyme is also known as acid maltase and is either completely absent or has decreased activity. This prevents the breakdown of the sugar storage form glycogen in the muscles. Glycogen is therefore stored in the muscle cells of the lysosomes, where it destroys the muscle cells. The remaining activity of the enzyme correlates with the severity of the disease.
The infantile type of Pompe disease has activity of less than one percent. The juvenile type retains a residual activity of up to ten percent and the adult type retains a residual activity of up to 40 percent. The disease is subject to autosomal recessive inheritance. Boys are affected just as often as girls. The causal gene defect was located in the q25.2-q25.3 region of chromosome 17 and is 28 kbp long.
The disease is genetically heterogeneous and has so far been associated with 150 different mutations. The patients are compound heterozygous. The infantile form is often characterized by two severe mutations. With this form, there is a high degree of correspondence between the genotype and the course of the disease. This is not the case with the adult form. The prevalence is given with values between 1: 40,000 and 1: 150,000. Around 200 people have been diagnosed in Germany.
Symptoms, ailments & signs
The main symptoms of Pompe disease are cardiomegaly and heart failure. There are also neurological and muscular deficits. The outbreak of Pompe disease is not limited to a certain age, but can affect all age groups. The infantile form occurs in infants and usually ends fatally in the first year of life. In most cases, the death that occurs is death from heart failure, which is due to hypertrophic cardiomegaly.
In the infantile form, the first symptoms appear after two months. Adolescents develop juvenile Pompe disease. In adults, medicine speaks of adult Pompe disease. These forms manifest themselves symptomatically in progressive muscle weakness in the respiratory muscles and the skeletal muscles of the trunk. The upper arms can be affected as well as the pelvis and thighs. The course is unpredictable in the adult and juvenile forms. Difficult courses are characterized by a loss of breathability.
There is also often a loss of mobility. States of exhaustion arise. In some cases, substances are deposited in the arteries and can form an aneurysm, the rupture of which can be fatal. On average, the first symptoms become noticeable shortly before the age of 30 and correspond to difficulties in running or exercising. The diagnosis is usually made in the mid-30s. Around ten years later, those affected are mostly dependent on a wheelchair.
Diagnosis & course of disease
The diagnosis of Pompe disease is usually confirmed by a muscle biopsy based on the anamnesis. Histologically, the massive glycogen deposits in the muscle can be demonstrated in the PAS staining. The diagnosis can just as easily be anchored in an enzyme activity measurement of the acid maltase, as can be detected in the leukocytes with the dry blood test. Molecular genetic examinations can also be used as diagnostic means. CK, CKMB, LDH, GOT and GPT are increased in the blood. The urine is usually elevated in Glc4.
Numerous differential diagnoses must be ruled out. In the infantile form, because of the pronounced symptoms, the suspected diagnosis can often be secured by the rapid progression that is associated with increasing breathing problems and motor development delay. Cardiomegaly can be confirmed by x-ray findings. Theoretically, prenatal diagnosis based on amniotic fluid examination or tissue removal is also conceivable.
The course of Pompe disease is usually more severe the earlier the disease breaks out. Nonetheless, Pompe's disease is characterized by an individual and therefore actually unpredictable course of the disease. Mild forms are also observed.
Complications
With Pompe disease, those affected suffer from significant restrictions and complaints in everyday life. In the first place, there are breathing difficulties, which lead to a greatly reduced resilience and fatigue of the patient. Furthermore, the lack of oxygen can also lead to a loss of consciousness, in which the person concerned can possibly injure himself from a fall.
The breathing difficulties also have a negative effect on the heart and the other internal organs, so that irreversible consequential damage can occur to the organs. Life expectancy is significantly reduced and restricted by Pompe disease. In the worst case, the affected person can die of cardiac death. Exhausting activities or sporting activities are still not possible for the person affected by this disease.
Treatment for Pompe disease is usually based on the symptoms and aims to increase life expectancy. Various drugs are used, which usually do not lead to particular complications. The symptoms can also be reduced with the help of physiotherapy. If there are psychological limitations, further psychological treatment is necessary.
When should you go to the doctor?
Anyone who suffers from Pompe disease is either affected by the hereditary disease as a child or has severe muscle symptoms as an adult. People with type II glycogenosis suffer from increasing muscle loss. The respiratory muscles can also be affected. If the genetic defect is not discovered early in a routine examination, visits to the doctor are the order of the day as the symptoms increase.
However, it can take a long time to get the diagnosis. First, there are many diseases with a similar course. Second, genetic testing is not the norm in medicine. Third, type II glycogen storage disease is also a metabolic disease. Many doctors are not familiar with the symptoms of Pompe disease.In addition, there is no uniform complaint for late-onset Pompe disease. The disease is much easier to diagnose in children.
Most adult patients see a doctor with a variety of muscular complaints. The symptoms described can affect the extremities, but also the respiratory muscles or the heart. Organs like the liver can also be affected. The decline is fast moving. As a result, over time, visits to the doctor increase without the correct diagnosis being made. An odyssey through the practices is not uncommon for Pompe disease.
Therapy & Treatment
Pompe disease has not yet been cured. A causal treatment of the symptoms is not available. Therefore, the patients are mainly treated symptomatically and supportively. Palliative forms of therapy are recommended. This therapy includes diet recommendations and breathing exercises as well as physiotherapy to strengthen the muscles. In the course of this, ventilation and artificial nutrition become necessary.
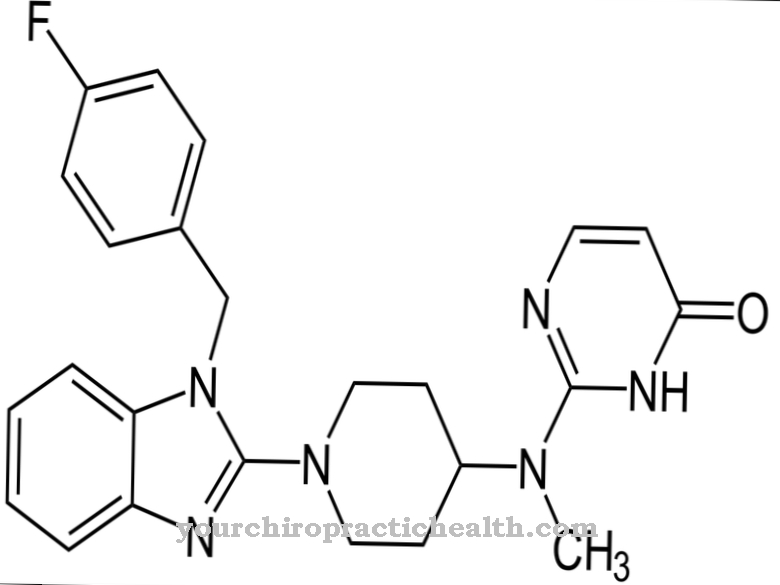
The timely decision for these measures is absolutely necessary to extend life. In terms of nutrition, a high-protein diet in combination with endurance training has proven itself. Since 2006, it has been possible to artificially supply the recombinant protein, which consists of CHO cells and is known as alglucosidase alfa or myozyme. The medication is administered intravenously every 14 days. Breakthrough successes were observed in infants after early administration of the drug, which could ensure survival.
There are contradicting experiences with older children and there is no convincing evidence for the effectiveness of the adult form. The cost of medication can be up to 50,000 euros per year for an infant and up to 500,000 euros per year for an adult. A lifelong supply is necessary. The response of the skeletal muscles to therapy is variable. But the heart muscle weakness improves. Because of the blood-brain barrier, the drug has no effect on disease processes in the brain.
Therapeutic approaches such as gene therapy are in the animal experimental stages. Gene transfer has already been successful in mice. Treatment with pharmacological chaperones can increase the residual activity of the acid maltase, but so far it cannot be used in a practical manner. Supportive psychotherapeutic care to deal with the situation is recommended for affected families.
You can find your medication here
➔ Medicines for painOutlook & forecast
Pompe disease is an incurable hereditary disease that usually results in a shorter life expectancy. The exact prognosis depends on the specific form of the disease and the age of the person affected when the disease breaks out.
The prognosis is worst for the early form of Pompe disease, which, if left untreated, usually leads to death within two years. Those affected mostly die of pneumonia or heart failure. Treatment of the disease with enzyme replacement therapy improves the prognosis significantly. The life expectancy of those affected increases significantly to an age of over ten years. Since this form of therapy is new, there are still no long-term prognoses.
The juvenile form of Pompe disease, if left untreated, usually leads to death before reaching adulthood. The adult form of Pompe disease has the most favorable prognosis. In any case, the life expectancy increases significantly with treatment. However, those affected usually develop some limitations such as cognitive difficulties and hearing loss or even deafness. New treatment options such as gene therapies for Pompe disease are currently being developed and tested, which should lead to a far more favorable prognosis.
prevention
So far, Pompe disease can only be prevented through genetic counseling during the family planning phase. For the parents of an affected child, the risk of recurrence is 25 percent. After a positive prenatal diagnosis, expectant parents also have the option of an abortion.
Aftercare
As a rule, those affected with Pompe disease have very few and only very limited measures and options for follow-up care available, so that they should ideally consult a doctor at an early stage in order to prevent other complaints and complications from occurring. It cannot cure independently, so that a doctor should be consulted at the first signs and symptoms of the disease.
The recovery process is primarily based on exercises from physiotherapy or physiotherapy. Many of the exercises can be repeated in your own home, which significantly promotes the treatment. The help and care of one's own family is also very important, which can also prevent depression and other psychological upsets.
In the case of an existing desire to have children, those affected should take advantage of genetic testing and counseling in order to prevent the disease from occurring again. Regular checks and examinations by a doctor are also necessary in the course of life. The disease itself usually does not reduce the patient's life expectancy. Further follow-up measures are usually not available to the person concerned.
You can do that yourself
For those affected, Pompe disease is a very stressful diagnosis, especially if the disease only manifests itself in old age. The disease can take a very individual course, which is why patients want to do as much as possible for a mild course of the disease.
The main thing is to prevent additional diseases. For example, patients should make sure that they are well supplied with oxygen so that they do not fall due to dizziness or suffer an accident. In addition, an insufficient supply of oxygen would damage the heart - and possibly other organs as well. Infectious diseases such as the flu or flu-like infections should also be avoided if possible because they also impair breathing and severely weaken the body and its immune system. Patients with Pompe disease would therefore do well to pay special attention to their immune system. You should ensure that you eat a fresh, high-protein diet as part of the diet recommendations of your specialist.
Regular exercise is also important to maintain muscle strength, especially in the legs. Above all, endurance training has proven itself in the treatment of Pompe disease. The treating physiotherapists provide the necessary assistance. Psychological support is helpful for many patients and their relatives. You can also get support and information from Pompe Deutschland e.V. (www.morbus-pompe.de).
.jpg)
.jpg)

.jpg)



.jpg)
.jpg)
.jpg)



.jpg)