Tyrosinemia are characterized by increased blood concentrations of the amino acid tyrosine. All forms of the disease have genetic causes. Type I tyrosinemia in particular leads to premature death if left untreated.
What is tyrosinemia?
In the Tyrosinemia is a genetic breakdown of the amino acid tyrosine, which leads to an increase in the tyrosine concentration in the blood. To date, three different forms of tyrosinemia are known. Causally, they differ in the location of the disturbance in the breakdown of tyrosine. All three forms of tyrosinemia are characterized by an increased tyrosine and phenylalanine concentration to different degrees:
- In tyrosinemia type I, toxic breakdown products are also formed in the body because the normal breakdown path is blocked by an enzyme defect at the end of the breakdown chain. These defective products of the tyrosine breakdown are toxic to the liver and kidneys, so that type I tyrosinemia is particularly severe.
- Type II tyrosinemia is mainly characterized by increased tyrosine and phenylalanine concentrations with all of their effects on the eyes, skin and nervous system. Here the breakdown of tyrosine is blocked at the beginning of the breakdown chain.
- The mildest and rarest form of tyrosinemia is type III tyrosinemia. The tyrosine and phenalalanine concentrations are less increased here. However, the increased concentrations have effects on the nervous system. In general, tyrosinemia is very rare. Type I tyrosinemia affects one to two per 100,000 people. There are only a few described cases of type III tyrosinemia.
causes
The general cause of all three forms of tyrosinemia is the disruption of the breakdown of tyrosine by defective enzymes. The form of the disease depends on the affected enzymes in the tyrosine breakdown chain. All tyrosinemias are caused by autosomal recessive mutations.
In type I tyrosinemia, the enzyme fumarylacetoacetate hydrolase is largely inoperable. Its coding gene is on chromosome 15. This enzyme is responsible for the last step in the tyrosine breakdown chain. The intermediates fumarylacetoacetate and maleyl acetoacetate are normally degraded in this reaction step.
However, if the enzyme is defective, these metabolites accumulate and are then converted into succinylacetoacetate and succinylcholine in an alternative reaction. However, these substances are strong liver and kidney toxins. Depending on how high their concentration in the blood is, they lead either quickly or through a chronic process to the complete destruction of the liver and kidneys.
Type II tyrosinemia is caused by a defect in the enzyme tyrosine aminotransferase. This enzyme initiates the first step in the breakdown of tyrosine. If it fails, tyrosine accumulates more and more in the blood. The concentration can be increased up to ten times the normal value. Since tyrosine is formed from the amino acid phenylalanine, the phenylalanine concentration also increases at the same time. Elevated phenylalanine concentrations are known to damage the nervous system.
At the same time, the eyes and skin are attacked by the high tyrosine levels. Finally, type III tyrosinemia is caused by a defect in the enzyme 4-hydroxyphenylpyruvate dioxygenase. The tyrosine and phenylalanine levels are only slightly increased here. Due to the blockage in the tyrosine breakdown chain, a backlog of tyrosine develops in all three forms of tyrosinemia, which is of course more pronounced the closer it is to the start of the breakdown chain.
Symptoms, ailments & signs
Type I tyrosinemia is characterized by damage to the liver, kidneys and brain. The disease manifests itself in newborns as poor drinking, vomiting, liver disease and kidney failure. There are two forms of the disease, both of which, if left untreated, lead to premature death from liver and kidney failure.
In the fulminant form, liver enlargement, edema and severe growth disorders occur early on. Death occurs within a few months of birth. In the milder form, the liver and kidneys are chronically degraded. Liver cirrhosis develops over a long process, which often leads to liver cancer.
If left untreated, death occurs by the age of ten at the latest. In tyrosinemia type II, damage to the cornea in the eyes, blistering and crust formation on the skin, and various neurological deficits occur. Type III tyrosinemia is characterized by mild mental impairment, impaired movement coordination and epileptic seizures.
Diagnosis & course of disease
Tyrosinemia can be diagnosed through various blood and urine tests. Increased tyrosine levels are found in urine samples. In addition, toxic metabolites such as succinylacetone can also be detected in the urine in tyrosinemia type I.
Complications
Depending on the type, tyrosinemia can cause various complications. Tyrosinemia type I can, due to the congenital liver, kidney and brain damage, cause symptoms such as poor drinking, liver diseases and renal insufficiency. The inability to drink can lead to dehydration and, as a result, to dehydration relatively quickly.
Liver disease always has serious effects on the entire body and can, for example, cause jaundice and severe inflammation of the internal organs. Renal insufficiency is just as serious because if left untreated, it can lead to kidney failure and thereby death. In the fulminant form, tyrosinemia can also promote growth disorders, edema and liver cancer as well as liver cirrhosis.
Type II tyrosinemia is associated with corneal damage, neurological deficits and other complications. Tyrosinemia type III can cause epileptic seizures, disorders of movement coordination and mental impairment in the course of the disease. When treating the breakdown disorder, the complications depend on the particular measure and the constitution of the patient.
The typically prescribed nitisinones can cause migraines and other side effects. A liver transplant always carries the risk that the body will reject the organ. Infections and wound healing disorders can also occur.
When should you go to the doctor?
The person concerned should always consult a doctor with tyrosinemia in order to prevent further complications or upsets. Early detection and subsequent treatment is very important so that the person affected should consult a doctor at the first signs and symptoms of the disease. In the worst case, the child can die of tyrosinemia. The doctor should be contacted for this disease if the child suffers from severe jaundice or diarrhea. Internal bleeding can also indicate this disease. There is also poisoning of the liver and other internal organs.
An increased heart rate or abnormal sensations in different parts of the body often point to the disease and should be checked by a doctor. Paralysis can develop anywhere on the body. Tyrosinemia should be treated promptly by a pediatrician or in a hospital. The further course depends on the time of diagnosis, so that no general prediction can be made. This disease may also reduce the child's life expectancy.
Treatment & Therapy
All forms of tyrosinemia are positively influenced by a diet low in tyrosine and phenylalanine. Such a diet can reliably improve the symptoms of tyrosinemia types II and III. However, Type I tyrosinemia is much more difficult to treat. In addition to a strict diet, the formation of toxic metabolites must also be prevented.
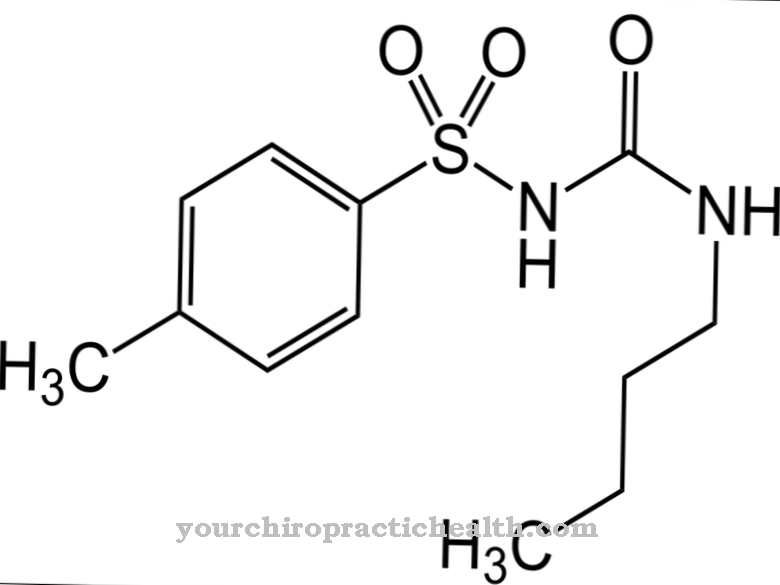
This can be achieved with the drug nitisinone (NTBC) by blocking an earlier breakdown step. This increases the tyrosine concentration in the blood. However, this can be kept low by the diet. Liver transplantation must be considered in advanced hepatic insufficiency.
prevention
Since the tyrosinemia is genetic, it cannot be prevented. However, through a strict diet low in tyrosine and phenylalanine, at least patients with tyrosinemia types II and III are able to lead a largely normal life. In patients with type I tyrosinemia, the concentration of the metabolites as well as of tyrosine and phenylalanine must be regulated for life through drug treatment and strict diet.
Aftercare
Tyrosinemia is a hereditary metabolic disorder. It is considered a rare disease and is classified using three forms I, II, and III. The options for treatment depend on the respective form. Appropriate follow-up care is necessary to achieve a favorable prognosis. The patient should be able to lead as unrestricted a life as possible.
In type II tyrosinemia, dietary treatment is often sufficient. However, the doctor's orders must be followed exactly. The healing process is checked during the follow-up and is set in the medium to long term. Tyrosinemia type III is the rarest form of metabolic disease. It is associated with a slight mental impairment and epilepsy.
During aftercare, those affected and their relatives learn how to deal with the disease on a daily basis. Special follow-up care is necessary for complete type I disease. Untreated, this tyrosinemia can be life-threatening. Internal organs such as the kidneys or the brain are damaged. An unfavorable course requires lifelong follow-up care.
Organ transplants may be an option at the doctor's discretion. It will be considered when other measures no longer help. Follow-up care takes place in the clinic, the patient's condition is closely monitored. Regular checks provide information on the compatibility of the new organ. Rejection reactions of the body must be avoided.
You can do that yourself
Tyrosinemia patients may adopt various dietary measures to support conservative management, depending on the type of disease. An energy-rich diet is important for type I tyrosinemia. The diet should allow as little tyrosine as possible to develop in the body. Catabolic situations, such as those that occur after a long period of hunger, should be avoided by eating meals regularly. The consumption of milk, egg and meat products must be severely restricted. Diet can slow the progression of the disease. It should be drawn up together with a doctor and a nutritionist and implemented consistently.
Tyrosinemia type II can also be treated supportively with an adapted diet. In type III tyrosinemia, in addition to dietary measures, preparations for medical emergencies must be made. In the event of an epileptic seizure, first aid measures should be initiated by calming the patient down and administering emergency medication. The person affected must be brought into a stable side position so that they do not injure themselves on objects or fall and also prevent vomit from entering the windpipe. The possible consequences of ataxia can be prevented through physiotherapy. Stairs, thresholds and dangerous objects in the household must be secured in order to minimize the risk of injury from falls. The exact measures for type I, II and III tyrosinemia should be discussed with a specialist.
.jpg)

.jpg)



.jpg)
.jpg)
.jpg)



.jpg)