Of the Von Willebrand factor is a protein that plays a crucial role in blood clotting. In the case of a deficiency in the coagulation factor, insatiable bleeding occurs.
What is the Von Willebrand Factor?
The Von Willebrand factor was named after the Finnish internist Erik Adolf von Willebrand. In his Swedish work Heredität pseudohemofili he described the clinical picture of a hereditary blood coagulation disorder. This was later named Von Willebrand Syndrome after him.
It wasn't until the 1950s that it was discovered that a deficiency in a protein that shortens bleeding time was the cause of Von Willebrand syndrome. This protein was then called Von Willebrand factor.
The Von Willebrand factor has a direct effect on hemostasis. Its direct effect is limited exclusively to cellular hemostasis, but plasmatic blood coagulation is also influenced. If there is a deficiency in Von Willebrand factor, hemostasis is impaired. Von Willebrand disease, often referred to as Willebrand-Jurgens syndrome, is the most common inherited hemophilia worldwide. An estimated 800 out of 100,000 people are affected. However, only two percent have significant symptoms.
Function, effect & tasks
Von Willebrand factor is the carrier protein for blood coagulation factor VIII. Coagulation factor VIII is antihemophilic globulin A. Von Willebrand factor circulates in the blood together with factor VIII. The formation of a complex protects the coagulation factor from proteolysis, i.e. from the breakdown of proteins.
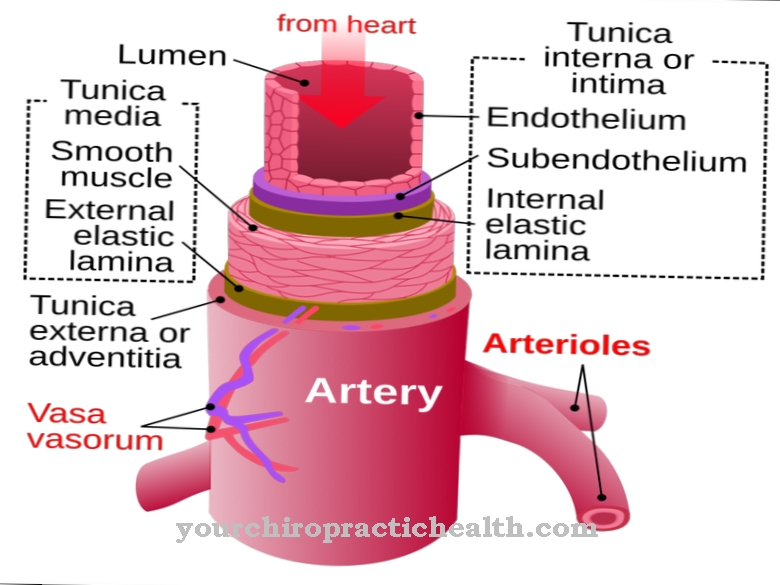
In the body, the Von Willebrand factor can bind to the Von Willebrand receptor. This receptor, which consists of the glycoprotein Ib / IB, sits on the surfaces of the blood platelets (thrombocytes). The Von Willebrand factor can also adhere to the proteins of the so-called subendothelial matrix. The subendothelial matrix is located directly below half the top layer of the vascular lining. In the event of injuries, the Von Willebrand factor can thus attach itself to the proteins or to the platelets. It acts as an adhesive protein and creates a connection between the platelets and the injury.
As a result, the Von Willebrand factor activates primary hemostasis. The platelets adhere to the fibers of the injured vessel wall and thus form a thin network over the wound. The platelets then release various substances which, by means of chemotaxis, attract more platelets. At the same time, these substances ensure that the affected blood vessel narrows and less blood can escape. The activated platelets accumulate and form a plug that temporarily closes the wound. This process of first stopping bleeding is known as cellular or primary hemostasis.
Education, occurrence, properties & optimal values
Von Willebrand factor is produced by the megakaryocytes and the endothelial cells of the inner wall of the blood vessels. Megakaryocytes are giant cells that are mainly found in the bone marrow. They are the precursor cells of the blood platelets. The platelets are constrictions of the megakaryocytes. They contain the Von Willebrand factor in their α-granules.
The Von Willebrand factor system is measured in the blood with various values together with the values of factor VIII. The term vWF: Ag describes the large-molecular and multimeric part of the system. This proportion can be understood as the actual Von Willebrand factor. In addition, for example, the vWF activity can be determined.
The differentiation of the individual components plays a role in the diagnosis of diseases in which parts of the Von Willebrand factor system are impaired. The reference value is 70-150% of the norm. The value depends on the blood group. The plasma concentration should be between 5 and 10 micrograms per liter.
Diseases & Disorders
Elevated levels of the Von Willebrand factor can be found in inflammation. The factor is a so-called acute phase protein. These proteins localize inflammation, prevent it from spreading and support the body's own defense system in the rehabilitation process.
The Von Willebrand factor in the blood can also be increased in rheumatic diseases, autoimmune diseases and cancer. Furthermore, taking the "birth control pill" can drive the value up. Decreased values are an indication of the presence of Von Willebrand-Jürgens syndrome.
This common blood clotting disorder is associated with an increased tendency to bleed. This is why Von Willebrand-Jürgens syndrome is one of the hemorrhagic diatheses. In the majority of cases, the cause of the disease is a hereditary disorder of the Von Willebrand factor system. The disease can be divided into different types. Type 1 has a quantitative factor deficiency. 80 percent of those affected belong to this group. They usually show rather mild symptoms. Long-lasting bleeding can occur, especially after operations. In women, menstruation is increased and large-area hematomas form in the event of impact injuries.
In type 2, there is sufficient Von Willebrand factor, but it is not fully functional. It is therefore a qualitative defect. Type 3 is the rarest form. Type 3 patients also show the most severe course. The Von Willebrand factor is completely absent in type 3 or is reduced to less than 5 percent.
The increased bleeding tendency leads to frequent nosebleeds (epistaxis), large areas of "bruising", long bleeding even after minor operations, increased menstrual bleeding and bleeding in the joints (hemarthrosis). Most patients with Von Willebrand-Jürgens syndrome do not require long-term therapy. However, patients should avoid drugs containing acetylsalicylic acid. These also inhibit the function of the platelets. If there is frequent nosebleeds, vasoconstricting nasal sprays can be used.
The increased menstrual period can be treated with hormonal contraceptives with a higher proportion of progestin. With type 3, these measures are not sufficient. In most cases, the trauma factor is substituted here. A prophylactic substitution at intervals of two to five days is also possible.






.jpg)
.jpg)
.jpg)



.jpg)