The Enzyme replacement therapy is used to treat lysosomal storage diseases, in which the lack of enzymes leads to a pathological accumulation of degradation products in the lysosomes of the cells.
The missing enzymes due to genetic defects are compensated for by regular intravenous infusions. Because the infused synthetic enzymes cannot cross the blood-brain barrier due to their molecular size, the therapy only works for lysosomal storage diseases that do not affect the central nervous system.
What is enzyme replacement therapy?

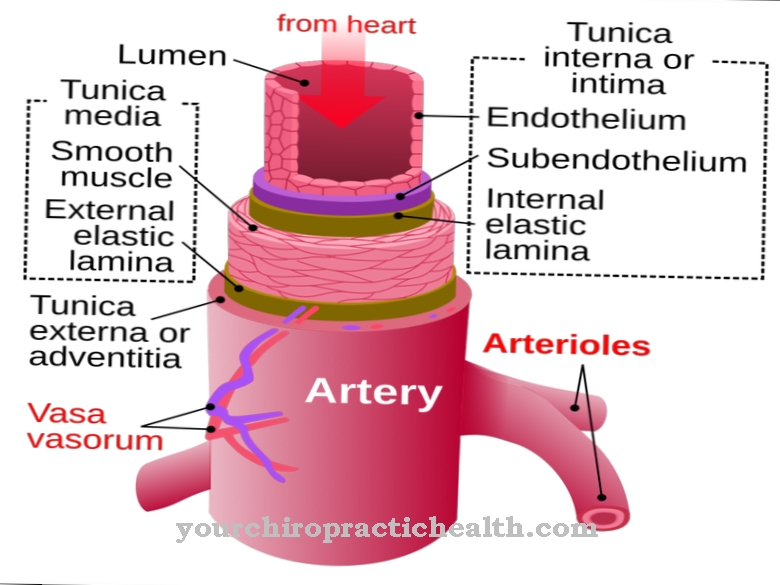
Lysosomes are special cell organelles in which foreign and endogenous substances are broken down and partially recycled. Specific hydrolyzing enzymes are required for the degradation and transport of the substances. These are proteases, nucleases, lipases and transporter substances.
A number of known genetic defects can lead to a failure of certain enzymes, so that some degradation products accumulate in the lysosomes in pathological quantities and accumulate until they reach the extracellular matrix, i.e. the intercellular spaces, in an uncontrolled manner. All genetic defects that lead to the failure of at least one necessary hydrolase are summarized under the term lysosomal storage disease. The enzyme replacement therapy (ERT, enzyme replacement therapy) is used to replace the missing endogenous enzymes with synthetically produced enzymes.
Because hydrolases are made up of relatively large molecules, they cannot be absorbed from the intestine without first being broken down and inactivated, so that they can only be administered via intravenous infusion. However, the size of the enzyme molecules also prevents the blood-brain barrier from being crossed, so that the therapy can only be effective for lysosomal storage diseases that do not affect the central nervous system (CNS).
Function, effect & goals
Over 50 different lysosomal metabolic disorders are known, each of which can be traced back to a monogenetic defect. The lysosomal storage diseases can be divided into seven different classes depending on the excessively stored substances due to the existing enzyme defect.
Mucopolysaccharidoses and oligosaccharidoses are primarily suitable for an ERT. The aim of ERT is always to compensate for the specific enzyme deficiency by means of the artificially supplied enzymes in order to bring the disease to a standstill or at least a milder course. In detail, replacement enzymes are available for the following lysosomal storage diseases:
- Gaucher's disease
- Pompe disease
- Fabry disease
- Hurler-Pfaundler syndrome (mucopolysaccharidosis I)
- Hunter's disease (mucopolysaccharidosis II)
• Maroteaux-Lamy syndrome (mucopolysaccharidosis VI) • Niemann-Pick B
Gaucher's disease is the most common lysosomal storage disease. It occurs in three different variants, two of which also affect the nervous system. In the non-neuropathic form, the spleen is particularly affected, which greatly enlarges and leads to secondary damage such as anemia and damage to the bone marrow. Typical symptoms are bone and joint pain and circulatory disorders. The acute neuropathic variant of the disease shows a severe course and offers little chance of survival beyond the first two years of life.
The storage disease Pompe disease is due to a deficiency of the enzyme alpha-1,4-glucosidase, which is involved in a large number of metabolic processes. Pompe disease leads to an enormous enlargement of the heart (cardiomegaly) and heart failure. There are early, serious, courses that appear in the first few months of life, as well as milder forms that only appear in later years of life.
Fabry disease is caused by an X-linked genetic defect, so only boys and men can be affected by the storage disease. The disease usually leads to symptoms in advanced childhood, including pain attacks, keratomas of the skin, kidney problems and cardiac muscle damage. The deficiency of the enzyme alpha-galactosidase A leads to an accumulation of ceramide trihexoside, which is the cause of the triggering of symptoms, which can also affect the autonomic nervous system.
It is not uncommon for the damage to lead to a heart attack, kidney infarction or even a stroke. The Hurler-Pfaundler syndrome is also known as mucopolysaccharidosis, type I and is caused by a disruption of the glycosaminoglycan metabolism. The disease is associated with a wide variety of symptoms, including severe mental impairment and severe skeletal changes. The course of the disease is severe, so that the average life expectancy is given as 11 to 14 years. Hunter's disease corresponds to mucopolysaccharidosis, type 2 and is - like Hurler's disease - caused by an X-linked defect. The disease is characterized by courses of varying severity, from occurring in early childhood to mild courses that only appear in adult men.
Due to the most common cardiac symptoms such as heart valve defects and heart muscle problems, life expectancy ranges from normal to slightly restricted. Maroteaux-Lamy syndrome (MPS VI) is one of the mucopolysaccharidoses that are inherited in an autosomal recessive manner because the causative gene defect is not on the X chromosome. The disease is very rare, with one case per 455,000 births. There are known mild and severe forms.
Symptoms are enlarged liver and spleen, carpal tunnel syndrome, and changes in the heart valves. The Niemann-Pick B is a sphingomyelin lipidosis, which is one of the lysosomal storage diseases and is caused by a genetic defect on chromosome 11. While type B of the disease mainly affects the liver and spleen, type A also has considerable neuronal problems.
You can find your medication here
➔ Medicines for painRisks, side effects & dangers
Since many of the lysosomal storage diseases that can be treated with enzyme replacement therapy take a severe course with a correspondingly increased mortality rate, the greatest risk in ERT is that the selected replacement enzyme does not work or works only too weakly.
Another risk lies less in the therapy itself than in the fact that the underlying disease is recognized too late, so that the ERT can stop during the course, but the damage that has already been caused cannot regress. About every second patient treated temporarily reacts to the infusions with symptoms such as fever and chills. The reasons for this are not yet fully understood. Some patients react by forming antibodies and there have been known cases where patients have reacted with rashes and bronchospasm.






.jpg)
.jpg)
.jpg)



.jpg)