The Hyperammonaemia is characterized by an increased concentration of ammonia in the blood. Congenital defects of the urea cycle and certain enzymes as well as severe liver diseases are possible causes. If left untreated, the disorder can lead to severe brain damage or even death.
What is hyperammonaemia?

© fadzeyeva - stock.adobe.com
Hyperammonaemia is the scientific term for an increased serum concentration of ammonia in the blood. Ammonia is produced during the breakdown of amino acids. In the so-called urea cycle, free ammonia is bound to form urea.
The non-toxic urea is then excreted in the urine. If, however, disturbances occur in the urea cycle due to defective enzymes, the ammonia formed can often not be converted into urea. Ammonia then accumulates in the blood and often causes irreversible damage, especially in the brain.
Any form of hyperammonaemia can produce the symptoms of hepatic encephalopathy. This disease is always the result of hyperammonaemia. Although hepatic encephalopathy is always described in the context of severe liver insufficiency, the hyperammonaemia that actually triggers it can also have other causes.
causes
There are many causes of hyperammonaemia. It is merely a symptom of an underlying disease. Often it is the result of a genetic disorder of the urea cycle. The urea cycle is controlled by several enzymes, the failure or failure of which can inhibit the synthesis of non-toxic urea from ammonia.
These enzymes include ornithine transcarbamylase, carbamoyl phosphate synthetase I, argininosuccinate synthase, argininosuccinate lyase, N-acetylglutamate synthetase (NAGS) and arginase 1. Ornithine transcarbamylase is most frequently affected. The ornithine transcarbamylase catalyzes the conversion of ornithine to citrulline.
If this reaction is interrupted, ammonia builds up in the blood. The failure of the other enzymes presented would also interfere with the breakdown of ammonia. These defects are a little less common. For example, carbamoyl phosphate synthetase I catalyzes the addition of ammonia, ATP and carbon dioxide to carbamoyl phosphate.
Argininosuccinate synthase is responsible for converting citrulline and aspartate into argininosuccinate. Argininosuccinate in turn serves as a starting material for the synthesis of arginine, which directly controls the formation of urea from ammonia. The enzyme argininosuccinate lyase catalyzes the breakdown of argininosuccinate into fumarate and arginine.
Arginase 1 controls the final step of the urea cycle with the breakdown of arginine into urea and ornithine. While the ornithine transcarbamylase defect is inherited in an x-linked manner, all other enzyme defects follow an autosomal recessive inheritance. There are also metabolic disorders outside of the urea cycle that can lead to hyperammonaemia.
These include organic acidurias, which cause organic acids to accumulate. These toxic metabolic intermediates, in turn, disrupt the urea cycle. Severe liver diseases are also possible secondary causes of hyperammonaemia, since ammonia is converted into urea in the liver.
Symptoms, ailments & signs
The symptoms of hyperammonaemia differ depending on the age of onset. In newborns, the course is life-threatening with the occurrence of poor drinking, hypotension and lethargy. If the disease only occurs in infancy, it is less acute with lethargy and failure to thrive.
When it first manifests in toddlerhood up to puberty, mental retardation, disorders of movement coordination, learning problems, headaches and vomiting are in the foreground. Overall, the appearance of hyperammonaemia corresponds to the symptoms of hepatic encephalopathy.
The hepatic encephalopathy described in connection with severe hepatic insufficiency manifests itself in a spectrum from mild clinical symptoms to coma. In the first stage, concentration disorders, mood swings, drowsiness and disorders of fine motor skills occur, among other things.
Stage II is characterized by increased drowsiness, speech motor disorders, apathy and disorientation. In stage III, the patient usually sleeps permanently, but can still be woken up. Disjointed awakening speech and increased muscle tension also belong to this stage. Stage IV is characterized by a hepatic coma (coma hepaticum).
Diagnosis & course of disease
Hyperammonaemia can be diagnosed by the appearance of symptoms of hepatic encephalopathy. Among other things, a skull CT is used for the differential diagnosis of a stroke or a blood sugar test to rule out hypoglycemia. Ammonia in the blood is also determined.
Complications
Without treatment, hyperammonaemia can cause serious harm to the patient, which in the worst case can even lead to death. The main result of hyperammonaemia is drinking weakness. This usually results in dehydration, which generally has a very negative effect on the patient's body.
Furthermore, mental retardation occurs, so that the person concerned may be dependent on the help of other people in everyday life. Thought processes are also restricted and made more difficult by the disease. Vomiting, nausea and headache occur. The coordination and all movements are also disturbed and can no longer be carried out easily.
The person may lose consciousness or even fall into a coma. Speech disorders and concentration disorders also occur. The quality of life is extremely limited by the complaints. If the hyperammonaemia is not treated, the life expectancy is reduced and the patient dies prematurely.
Treatment does not lead to further complications and is carried out with the help of drugs. The symptoms can be limited relatively well. However, it is possible that the hyperammonaemia has already caused irreversible damage.
When should you go to the doctor?
Newborns and young children who show refusal to eat should see a doctor as soon as possible. If there is a refusal to supply with breast milk or a substitute infant formula, there is cause for concern. A doctor is needed in the event of weight loss, pale skin or insufficient saliva production. In the further course, without medical care, there is a risk of an insufficient supply of the organism and thus the premature death of the newborn. If the child shows behavioral problems, is lethargic or if the movements of the limbs are uncoordinated, a doctor must be consulted.
If there are fluctuations in mood, severe tiredness and a very intensive need for sleep, a check-up should be carried out. If increased tension in the muscles is found, this must be checked medically. If the muscles cannot be loosened even when lying down or when sleeping lightly, a doctor should be consulted. If the child is comatose, call an ambulance as it is an emergency. If the child is noticed by disorders of fine motor skills or coordination problems, a doctor should be consulted. In the event of a loss of consciousness or apathy, a doctor must be consulted immediately. Children who can already speak must be presented to a doctor as soon as their pronunciation is subject to interruptions or the language ability declines.
Doctors & therapists in your area
Treatment & Therapy
In the event of acute hyperammonaemia, urgent measures must be taken. For this it is necessary to immediately stop the protein intake for two days. Furthermore, this includes performing a sugar infusion, administering insulin and adding arginine and carnitine.
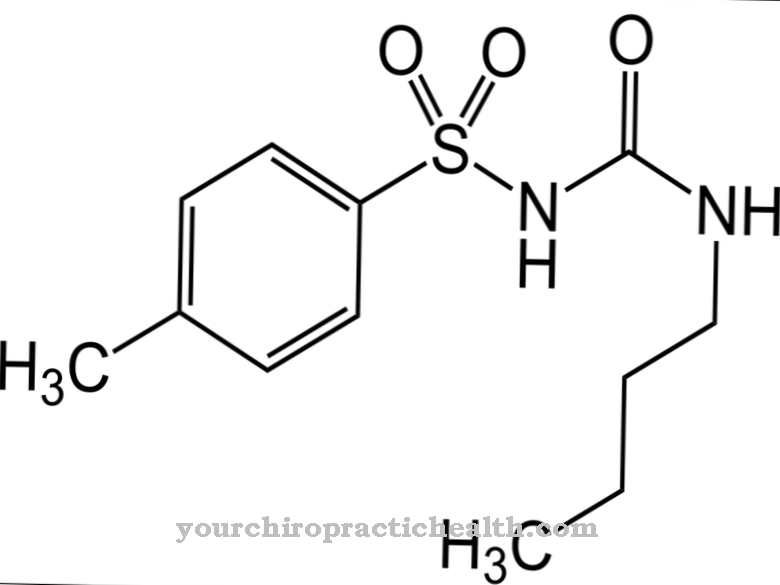
Various drugs such as phenyl acetate, phenyl butyrate or benzoate are used to detoxify the blood. Diuretics increase urine excretion. In some cases dialysis must also be performed. Lactulose, a disaccharide, is also given.
With the help of intestinal bacteria, this carbohydrate is broken down into lactate and acetone. The resulting acidic intestinal environment ensures the binding of ammonia to ammonium salts. For the long-term treatment of hyperammonaemia in the case of enzyme defects, a low-protein diet must be followed permanently.
In addition, the administration of arginine hydrochloride, citrulline or lysine is necessary. If the hyperammonaemia is the result of liver disease, it must of course be treated.
Outlook & forecast
The prospect of a cure for hyperammonaemia is linked to the use of treatment and the underlying disease. With drug therapy, the blood is detoxified. This alleviates the discomfort and improves the patient's health. If treatment is refused, the prognosis is poor. In severe cases, brain activity can be permanently impaired. In addition, the person concerned is threatened with premature death.
The prognosis improves with treatment, and there is usually no curability. The medical care initiated depends on the cause of the symptoms. Symptoms can be alleviated in the case of a genetic underlying disease. In long-term therapy, the urea cycle is monitored and regulated. This leads to an improvement in the general state of health and a decrease in the existing complaints. Since the genetic defect cannot be changed for legal reasons, there is no permanent cure. As soon as the treatment is interrupted or terminated at your own request, the disorders of the urea cycle return.
In an acute condition, the patient receives intensive medical care. This improves the health of the patient, but is not sufficient for a cure. If there is organ damage, the organism must also be given long-term medical support so that the blood can be adequately cleaned.
prevention
Serious crises in congenital hyperammonaemia can be avoided by following a lifelong diet with low-protein foods. However, there is no recommendation to prevent the disease if the urea cycle is disrupted because it is genetic.
In the event of a familial appearance, genetic counseling is offered. However, non-genetic causes of hyperammonaemia can be prevented through a healthy lifestyle and avoidance of alcohol.
Aftercare
As a rule, the person affected with hyperammonaemia has no or only very few measures and options for follow-up care available. The person concerned is primarily dependent on an early diagnosis with subsequent treatment so that there are no further complications or complaints. An early detection with a subsequent treatment always has a very positive effect on the course of the disease.
Hyperammonaemia may reduce life expectancy if the disease is recognized late or is not treated. Those affected are dependent on taking medication with hyperammonaemia. It is important to ensure that it is taken regularly and, above all, that it is taken correctly so that the symptoms can be treated correctly. If you have any questions or questions, you should always contact a doctor first.
Regular examinations of the body are also very important in this disease in order to prevent damage to the internal organs or to detect them early. A healthy lifestyle with a healthy diet can also have a positive effect on the course of the disease. In many cases, those affected by the disease are dependent on the care and help of relatives and families.
You can do that yourself
If there is acute hyperammonaemia, the protein intake must be stopped immediately. Depending on the medical treatment, the diet should then be changed to avoid a new reaction. The doctor will recommend a low-protein diet that should be maintained for at least two to three months after diagnosis.
At the same time, therapy using various detoxification drugs is indicated. This can be supported by the sick person through regular hydration. In addition, physical activity should be temporarily avoided. Rest and bed rest are important, especially in the first one to two weeks after diagnosis.
If the hyperammonaemia occurs as a result of a liver disease, stimulants must be avoided. In addition, the underlying disease must be treated. The underlying disease is usually treated with medication, but in some cases surgical treatment is necessary. After an operation on the liver, the body is very weak. The patient has to spend a few days in the hospital and should then cure himself at home. Regular visits to the doctor's office ensure close monitoring and targeted treatment of acute complaints.


.jpg)
.jpg)



.jpg)
.jpg)
.jpg)



.jpg)