in the Urea cycle metabolic end products containing nitrogen are converted to urea. This biochemical process takes place in the liver. The urea is then excreted through the kidneys.
What is the urea cycle?

Proteins, i.e. proteins, are made up of many amino acids. These in turn contain at least one nitrogen molecule in the form of an amino group (-NH2). If the amino acids with their nitrogen molecules are broken down, toxic ammonia (NH3) is produced. The ammonia is dissolved in the blood in the form of so-called ammonium ions (NH4 +). The substance can also have a toxic effect in this dissolved form. Urea is formed in the liver by binding ammonium ions. This renders the ions harmless. The resulting urea is excreted through the kidneys.
Humans depend on the urea cycle. Most aquatic animals can immediately release the ammonia they accumulate into the water via their body fluids via osmosis. In birds and lizards, the less harmful uric acid is produced instead of urea. Although this is also excreted in the urine, unlike urea, it can remain in the body longer without causing damage.
Function & task
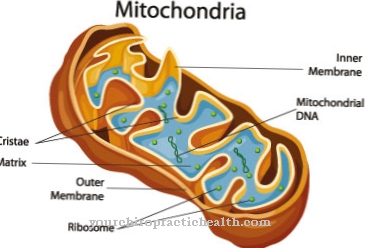
The urea cycle, too Ornithine cycle called, begins in the mitochondria. Mitochondria are also known as the power plants of the cell, as the very high-energy molecule ATP is produced here. Within the matrix of the mitochondria, the enzyme carbamoyl phosphate synthetase 1 forms carbamoyl phosphate from free ammonia and carbon dioxide.
A phosphate residue remains in this reaction. This is required in the next step. Here ornithine, an amino acid that is present in the mitochondrial matrix, reacts with the carbamoyl phosphate formed in the first step. The carbamoyl phosphate transfers its carbamoyl group to the ornithine. Citrulline and phosphate are formed. The catalyst of this chemical reaction is the enzyme ornithine transcarbamylase.
For the rest of the process, the resulting citrulline must be transported from the mitochondria into the cell fluid of the liver cells (hepatocytes). This is done using the ornithine-citrulline transporter. In the cytoplasm of the hepatocytes, the amino group aspartate also becomes part of the urea cycle. The carbonyl group of citrulline reacts with aspartate. The catalyzing enzyme arginine succinate synthetase produces arginino succinate. This is split into free furamate and free arginine by another catalyzing enzyme, argininosuccinase.
The free furamate is regenerated to aspartate. The arginine is in turn cleaved by the enzyme arginase. This creates urea and ornithine. The ornithine is transported back into the mitochondrion and serves as a carrier molecule for the formation of citrulline. The urea is excreted through the kidneys as a water-soluble molecule.
Without the urea cycle, the metabolic toxin ammonia cannot be disposed of. The urea is used to detoxify the body. If it is disturbed, it can lead to severe neurological symptoms.
A healthy liver is particularly important for a functioning urea cycle, as this is where most of the urea formation takes place. Only a small and negligible part of the urea formation is carried out in the kidney. However, since the kidneys excrete urea, the urea content in the blood is used to record and monitor the progression of renal insufficiency. The urea content in the blood also plays a role in dialysis monitoring or in determining a coma cause.
Illnesses & ailments
A total of six disorders of urea metabolism are known. These are always the result of a disruption of one of the enzymes involved. In the case of urea metabolism disorders, there is usually a deficiency of carbamoyl phosphate synthetase, ornithine transcarbamylase, argininosuccinate synthetase, argininosuccinate lyase, arginase or N-acetylglutamate synthetase. A deficiency in one of these enzymes always leads to a pathologically high accumulation of ammonia in tissue and blood.
An increased ammonia level in the blood is also called hyperammonaemia. Hyperammonaemia can also be caused by abnormal liver functions. In particular, advanced liver diseases such as chronic hepatitis or liver cirrhosis impair the urea cycle through the destruction of liver cells.
The main consequences of a severe disruption in the urea cycle are damage to the central nervous system. These symptoms are also referred to as hepatic encephalopathy. If the urea cycle is disturbed, too much toxic ammonia remains in the blood. The cell poison mainly attacks the cells of the nervous system. These swell due to the poisoning. This increases intracranial pressure and ultimately brain edema occurs.
The symptoms can be divided into four levels. In the first stage there are only slight changes such as concentration disorders or mood swings. In some cases, however, those affected already have problems to solve easy arithmetic tasks at this stage. In the second stage, there is increased drowsiness. The time orientation is limited. This is followed by speech and consciousness disorders. Patients experience abnormal drowsiness but are still responsive and awakenable. The most severe form of hepatic encephalopathy is hepatic coma, also known as coma hepaticum. This stage is characterized by complete loss of consciousness and a complete lack of reflexes. The hepatic coma is often fatal.
The manifestation of symptoms in disorders of the urea cycle is favored by several factors. Infections can lead to increased cell decay and thus to an increased accumulation of amino acids. An increased intake of proteins with food can overwhelm the already disturbed urea cycle.
The therapy of disorders in the urea cycle is medication with phenyl acetate and benzoate. Both react together with glutamine and glycine to form phenacetylglutamine and hippuric acid. Like urea, these can remove nitrogen and are also excreted in the urine.









.jpg)


.jpg)